There is a piece in Technology Review about 5G wireless.
The writer states:
Samsung says its new transceiver can send and receive data at speeds
of more than a gigabit per second over up to two kilometers—and it could
deliver tens of gigabits per second at shorter distances. This compares
to about 75 megabits per second for the latest standard, known as 4G
LTE. The Samsung technology relies on 28-gigahertz frequencies, which
can carry commensurately more data but can be blocked by buildings,
people, foliage, and even rainfall.
Samsung says it has greatly mitigated these problems by sending data
over any of 64 antennas, dynamically shaping how the signal is divided
up, and even controlling the direction in which it is sent, making
changes in tens of nanoseconds in response to changing conditions (among
other features, it can catch stray reflections of signals that had
bounced off an obstruction).
In my review of the Crawford book I indicated as such. This is just the first of many embodiments of Gbps to the user wireless. This is also why Verizon abandoned FIOS and why one should wonder about CATV.
It continually amazes me how regulators and lawyers are often clueless about technology and develop policies based upon bad technology and ignorance of what is doable. Well that is Washington after all.
Friday, May 31, 2013
PSA Issues Keep Returning
The Trustees of SSI and Medicare have just issued their
annual report on the health of that system[1].
Needless to say the system faces challenges, substantial challenges. The ACA
proposes various ways to manage them and for better or worse it is rationing.
Not a direct and blatant form of rationing but a more subtle and possibly
sinister form which is the result of “new” or “updated” clinical testing
regulations. At the heart of this is the PSA issue.
The AUA has issued a new set of guidelines which put
additional pressure on PSA testing[2].
The recommendations are as follows:
1. The Panel recommends against PSA screening in men
under age 40 years. (Recommendation; Evidence Strength Grade C) In this age
group there is a low prevalence of clinically detectable prostate cancer, no
evidence demonstrating benefit of screening and likely the same harms of
screening as in other age groups.
2. The Panel does not recommend routine screening in men
between ages 40 to 54 years at average risk. (Recommendation; Evidence Strength
Grade C) For men younger than age 55 years at higher risk (e.g. positive family
history or African American race), decisions regarding prostate cancer
screening should be individualized.
3. For men ages 55 to 69 years the Panel recognizes that
the decision to undergo PSA screening involves weighing the benefits of
preventing prostate cancer mortality in 1 man for every 1,000 men screened over
a decade against the known potential harms associated with screening and
treatment. For this reason, the Panel strongly recommends shared
decision-making for men age 55 to 69 years that are considering PSA screening,
and proceeding based on a man’s values and preferences. (Standard; Evidence Strength
Grade B) The greatest benefit of screening appears to be in men ages 55 to 69
years.
5. The Panel does not recommend routine PSA screening in
men age 70+ years or any man with less than a 10 to 15 year life expectancy.
(Recommendation; Evidence Strength Grade C) Some men age 70+ years who are in
excellent health may benefit from prostate cancer screening.
Simply stated the guideline limits testing to men 55 to 69
years of age. It does not recommend testing for men over 69 years of age, the
age at which most prostate cancer occurs.
Now the evidence used which I have reviewed in my work on
Prostate Cancer Genomics[3] demonstrates
the clear problems with the major studies, problems that many in the field have
recognized and thus in a similar fashion discount the study out of hand. Thus
the basis for these recommendations is highly tainted and suspect.
Now in a paper written for the magazine Columbia Medicine,
the alumni magazine for P&S grads and affiliates, the author states[4]:
The PSA controversy exploded in May 2012, when the U.S.
Preventive Services Task Force gave the test a resounding "D" grade,
recommending against its use as a screening tool. The independent panel of experts
in prevention and primary care (no urology or oncology experts were on the
panel) found that the test led to "overdiagnosis" of prostate cancer
and, consequently, overtreatment. For Dr. Benson, as for many urologists across
the United States, that line of thinking is unworkable on multiple levels. "There's
no such thing as overdiagnosis; there's only overtreatment," he says.
"And you can't decide whom to treat and whom not to treat, you can't
establish risk, without diagnosis. So the concept of overdiagnosis is
dangerous, and it will preclude patients from getting life-saving therapy when
it's indicated."
Dr Benson is head of Urology at Columbia University Medical
Center and a world expert in treating this disease. I know the group there
quite well and Benson and his team are truly world class. Benson is at the forefront
of the battle. The article continues:
Dr. Benson stresses that the overall boundaries are
clear. "The goal for prostate cancer treatment is death from something else,"
he says. "Prostate cancer is, in general, a slow growing cancer, so to
take an 85-year-old man with a PSA of less than 4 and say he needs ongoing PSA
screening is insane. But to take an 85-year-old man who has never had a PSA
ever in his life and say he shouldn't get one, just to see where he's at, is
also crazy. I think every man, regardless of age, deserves one PSA. If that one
PSA places you in a category where the chance of your dying of prostate cancer
is low, then you don't need to have biopsies and additional PSA testing. This
is a $15 blood test. The rub here is not in the PSA; the rub is in what people
do with the data."
Benson is also quoted as follows:
One of the department's most interesting areas of current
research is in confirmatory biopsies to decide eligibility for active
surveillance among men who come to Columbia after being diagnosed elsewhere.
"I have greater confidence in our ability to thoroughly biopsy the prostate,"
says Dr. Benson, who performs biopsies with 24 to 30 cores rather than the more
typical 12. To test the accuracy of initial biopsies at other facilities and
consequent eligibility of patients for active surveillance, Dr. Benson selected
60 incoming patients and, before enrolling them in active surveillance,
repeated their biopsies to be sure that the original tests hadn't missed
prostate cancer that might be of greater risk.
This is a critical difference, namely the increased core
density. We have demonstrated the probability of detection versus number of
cores, and also total prostate volume. Going from the old six cores to 24
dramatically increases the detection probability, especially is using
trans-rectal ultrasound guiding by ax experienced urologist. The difference
between a procedure at Columbia versus at some local clinic can be orders of
magnitude in terms of detection probability. Also the sequelae problems are
generally much lower, even with more cores.
Finally Benson states:
For Dr. Benson, his current touchstones show the way forward.
"There are two telling statistics," he says. "One is that before
PSA screening, the most common presentation was a patient with metastatic disease.
And the second thing is that metastatic disease is normally rare and the death
rate has been reduced by 40 percent. What we have to do is find ways of continuing
to have a death rate reduced by 40 percent while not treating people who don't
need treatment. That has to be the goal."
Eliminating a test which has a false alarm rate is clearly
NOT the way to go, it is especially not the way if we want an informed patient
as part of the decision process.
The problem here is that PSA measurements and other types of
non-invasive measurements are critical and especially if we have temporal data
for the patient, namely annual or even semi-annual PSA tests along with %Free
PSA, and we can calculate velocity and other measures, normalize them for Prostate
volume, then we have a substantially better test. Combine that with competent
and experienced urologists and biopsies then we can ascertain what the true
state of reality is.
Having some panel decide based on faulty data is NOT the way
to proceed.
[1] http://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/ReportsTrustFunds/Downloads/TR2013.pdf
[4] Baruchin,
A., To Test or Not to Test? To Treat or Wait and See? Columbia Medicine, Spring
2013, pp 20-25.
Wednesday, May 29, 2013
Two More Melanoma Therapeutics
The FDA just approved two more melanoma therapeutics.
They state:
The U.S. Food and Drug Administration today approved two new drugs, Tafinlar (dabrafenib) and Mekinist (trametinib), for patients with advanced (metastatic) or unresectable (cannot be removed by surgery) melanoma, the most dangerous type of skin cancer.
Tafinlar, a BRAF inhibitor, is approved to treat patients with melanoma whose tumors express the BRAF V600E gene mutation. Mekinist, a MEK inhibitor, is approved to treat patients whose tumors express the BRAF V600E or V600K gene mutations. Approximately half of melanomas arising in the skin have a BRAF gene mutation. Tafinlar and Mekinist are being approved as single agents, not as a combination treatment.
The FDA approved Tafinlar and Mekinist with a genetic test called the THxID BRAF test, a companion diagnostic that will help determine if a patient’s melanoma cells have the V600E or V600K mutation in the BRAF gene.
The FDA is rapidly issuing approvals on many of these therapeutics.
Cost and long term efficacy are still issues.
They state:
The U.S. Food and Drug Administration today approved two new drugs, Tafinlar (dabrafenib) and Mekinist (trametinib), for patients with advanced (metastatic) or unresectable (cannot be removed by surgery) melanoma, the most dangerous type of skin cancer.
Tafinlar, a BRAF inhibitor, is approved to treat patients with melanoma whose tumors express the BRAF V600E gene mutation. Mekinist, a MEK inhibitor, is approved to treat patients whose tumors express the BRAF V600E or V600K gene mutations. Approximately half of melanomas arising in the skin have a BRAF gene mutation. Tafinlar and Mekinist are being approved as single agents, not as a combination treatment.
The FDA approved Tafinlar and Mekinist with a genetic test called the THxID BRAF test, a companion diagnostic that will help determine if a patient’s melanoma cells have the V600E or V600K mutation in the BRAF gene.
The FDA is rapidly issuing approvals on many of these therapeutics.
Cost and long term efficacy are still issues.
Chaotic Data or You Just Do Not Know Enough
In a recent Scientific American piece the author bemoans the wealth of unorganized data available regarding cancers.
The author bemoans:
The field of genetics has flourished with the publishing of the complete human genome in 2001, aided by the advent of fast, affordable sequencing technology. A completed genetic code of a healthy person allows us to compare against the genetics of cancer. With advanced analytical techniques, and decades of research into the characteristics of different forms of this disease, it seemed that it was finally time to pull out the answers from the code itself by looking for the mutations that cause or support the cancer’s growth – the differences between the cancer cell and a normal cell. But when the answers didn’t bubble up from our statistics and reams of data, it became clear that the questions left for us were far more complicated.
Yes indeed the questions are complex. But we do have examples of previous work to guide us. Cancer is an amalgam of ligands, receptors, pathway elements, transcription factors, SNPs, miRNAs, and the ECM environment. Cancer cells move, mutate, and affect and are affected by their local environment. It is a stochastic distributed dynamic system. Fortunately we have been studying these for decades, if not for most of the past century. The issue is to understand what drives what and how to control it.
Cancer is a stochastic distributed temporally varying field, cells changing and flowing with ever changing genetic characteristics. We can understand these characteristics and even predict to some degree where they will go in the future. That is the challenge and the opportunity. The author seems to totally miss this point.
She continues:
One method for addressing chaos of this sort is to develop a statistical plan. We want to be able to take the information we know, look at the probabilities that certain changes will occur, and use statistics to determine what the cancer will likely do next. This challenge seems insurmountable when you look at the variables we must contend with: rapid evolution, unchecked growth, subtle migration, and so on. Changes that occur in the genome during cancer initiation and progression involve massive genetic rearrangements, damage, and mutation.
This makes it difficult to distinguish between causes and consequences of the cancer. It would be far easier if each gene, or even each chromosome, carried out its business without interaction with the rest of the cell. But not only can changes in one area of the genome have profound direct and indirect effects on the expression of genes elsewhere, we now have evidence that cancer cells can send these activity-altering signals to other cells, both tumor-derived and normal.
Frankly a "statistical plan" is the worst path to go. You most likely can correlate anything with anything. Economists do it all the time and look where they have taken us. We know of can know what affects what and how that occurs. We can and must stipulate the system relationships. We must have a system model NOT some set of correlations.
This I fear is the difference between the bench worker, the physician, and the engineer. Is cancer genetics ready for the engineer? I believe that the door for such an entry is now opening. Not everything is in place but we may have the key pieces to begin the journey.
We know causes, we understand the consequences. If BRAF V600 is present we even know how to inhibit it, for a while. We must then understand how to take the next step. Cell by cell we must understand the genetic progression as a system, not as a set of correlations.
The author bemoans:
The field of genetics has flourished with the publishing of the complete human genome in 2001, aided by the advent of fast, affordable sequencing technology. A completed genetic code of a healthy person allows us to compare against the genetics of cancer. With advanced analytical techniques, and decades of research into the characteristics of different forms of this disease, it seemed that it was finally time to pull out the answers from the code itself by looking for the mutations that cause or support the cancer’s growth – the differences between the cancer cell and a normal cell. But when the answers didn’t bubble up from our statistics and reams of data, it became clear that the questions left for us were far more complicated.
Yes indeed the questions are complex. But we do have examples of previous work to guide us. Cancer is an amalgam of ligands, receptors, pathway elements, transcription factors, SNPs, miRNAs, and the ECM environment. Cancer cells move, mutate, and affect and are affected by their local environment. It is a stochastic distributed dynamic system. Fortunately we have been studying these for decades, if not for most of the past century. The issue is to understand what drives what and how to control it.
Cancer is a stochastic distributed temporally varying field, cells changing and flowing with ever changing genetic characteristics. We can understand these characteristics and even predict to some degree where they will go in the future. That is the challenge and the opportunity. The author seems to totally miss this point.
She continues:
One method for addressing chaos of this sort is to develop a statistical plan. We want to be able to take the information we know, look at the probabilities that certain changes will occur, and use statistics to determine what the cancer will likely do next. This challenge seems insurmountable when you look at the variables we must contend with: rapid evolution, unchecked growth, subtle migration, and so on. Changes that occur in the genome during cancer initiation and progression involve massive genetic rearrangements, damage, and mutation.
This makes it difficult to distinguish between causes and consequences of the cancer. It would be far easier if each gene, or even each chromosome, carried out its business without interaction with the rest of the cell. But not only can changes in one area of the genome have profound direct and indirect effects on the expression of genes elsewhere, we now have evidence that cancer cells can send these activity-altering signals to other cells, both tumor-derived and normal.
Frankly a "statistical plan" is the worst path to go. You most likely can correlate anything with anything. Economists do it all the time and look where they have taken us. We know of can know what affects what and how that occurs. We can and must stipulate the system relationships. We must have a system model NOT some set of correlations.
This I fear is the difference between the bench worker, the physician, and the engineer. Is cancer genetics ready for the engineer? I believe that the door for such an entry is now opening. Not everything is in place but we may have the key pieces to begin the journey.
We know causes, we understand the consequences. If BRAF V600 is present we even know how to inhibit it, for a while. We must then understand how to take the next step. Cell by cell we must understand the genetic progression as a system, not as a set of correlations.
Tuesday, May 28, 2013
Weather or Climate
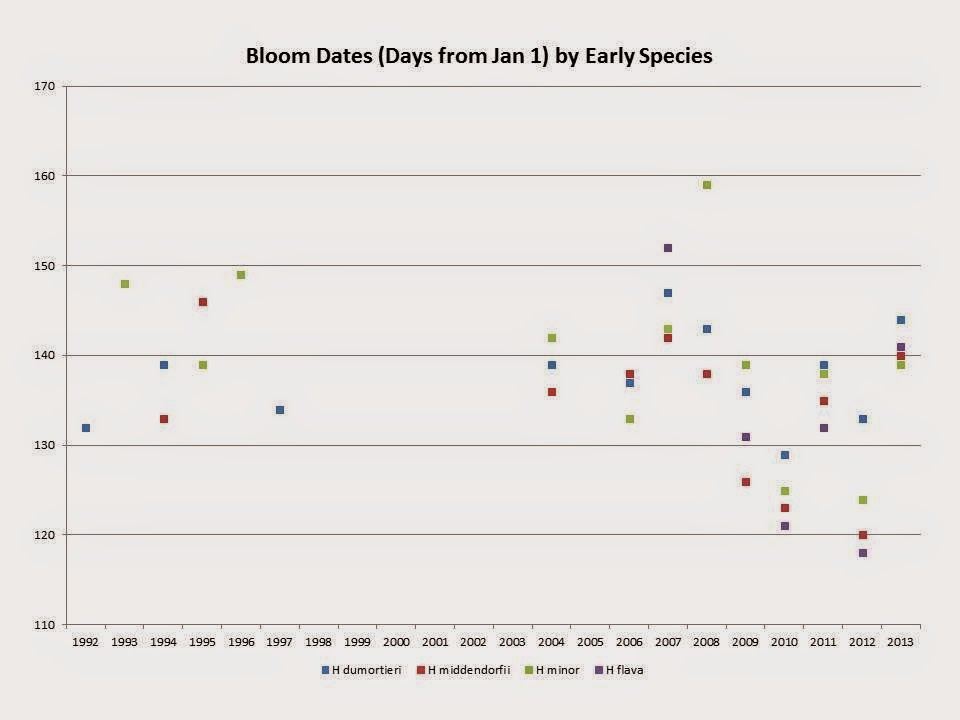



H middendorfii goes from 142 down to 131. That is a 9 day drop. It is generally one of the earliest. But look at the data, it is quite spread out.

But the question is; can we say that this is a trend or just normal variation? Trends would result in February blooms at some time which is doubtful. But it is interesting to always look at facts and species plants do tell us a great deal. The problem is the "noise" from any micro climate changes. For example the Hurricane got rid of many tall trees. Yet the flowers come out before any significant tree foliage. Thus perhaps we have a stable micro climate. Just a thought.
Monday, May 27, 2013
The Left and Taxes
We have laws, including Tax Laws. And we have Tax Lawyers. They are very expensive. My Tax Lawyer costs 4 times my Accountant. My Tax Lawyer is really important, my accountant makes sure I do everything right, but my Tax Lawyers makes sure the IRS understands that I did it right. I fear the IRS more than the Devil! As very well we all should.
But then again there are laws. If we follow them, then we have no problems. One of the Left Wing Economists located at Columbia, that school which denied Catholics admission because of their religion, not that I hold a grudge, states:
The world looked on agog as Tim Cook, the head of Apple, said his company had paid all the taxes owed – seeming to say that it paid all the taxes it should have paid. There is, of course, a big difference between the two. It's no surprise that a company with the resources and ingenuity of Apple would do what it could to avoid paying as much tax as it could within the law. While the supreme court, in its Citizens United case seems to have said that corporations are people, with all the rights attendant thereto, this legal fiction didn't endow corporations with a sense of moral responsibility; and they have the Plastic Man capacity to be everywhere and nowhere at the same time – to be everywhere when it comes to selling their products, and nowhere when it comes to reporting the profits derived from those sales.
With all due respect, there is no difference between the two. The laws says such and such and you then follow the law. What does this character mean by "should"? According to him? Who is he? The arrogance of the Left is amazing. If we do not like it then change the tax law.
Frankly we should eliminate all corporate taxes. Imagine what that would do! It would make all these companies move here, hire people here, rent and build facilities here. Then we can tax the result via personal taxes. After all we really do not get that much from companies. The big ones pay less as a percent of profits than the smaller ones. So "level the playing field", is that not the Socialist dictum, make the tax zero!
Besides why should we ever listen to a Socialist!
But then again there are laws. If we follow them, then we have no problems. One of the Left Wing Economists located at Columbia, that school which denied Catholics admission because of their religion, not that I hold a grudge, states:
The world looked on agog as Tim Cook, the head of Apple, said his company had paid all the taxes owed – seeming to say that it paid all the taxes it should have paid. There is, of course, a big difference between the two. It's no surprise that a company with the resources and ingenuity of Apple would do what it could to avoid paying as much tax as it could within the law. While the supreme court, in its Citizens United case seems to have said that corporations are people, with all the rights attendant thereto, this legal fiction didn't endow corporations with a sense of moral responsibility; and they have the Plastic Man capacity to be everywhere and nowhere at the same time – to be everywhere when it comes to selling their products, and nowhere when it comes to reporting the profits derived from those sales.
With all due respect, there is no difference between the two. The laws says such and such and you then follow the law. What does this character mean by "should"? According to him? Who is he? The arrogance of the Left is amazing. If we do not like it then change the tax law.
Frankly we should eliminate all corporate taxes. Imagine what that would do! It would make all these companies move here, hire people here, rent and build facilities here. Then we can tax the result via personal taxes. After all we really do not get that much from companies. The big ones pay less as a percent of profits than the smaller ones. So "level the playing field", is that not the Socialist dictum, make the tax zero!
Besides why should we ever listen to a Socialist!
Slow News Day - X Ray Scanners
Yes, X Ray scanners use X Rays. X- Rays cause cancer. Yes X Ray scanners penetrate only the top layers of the skin. Yes melanocytes reside there. Yes, X Rays mutate genes. Yes X Rays can mutate melanocyte genes. Yes mutated melanocytes can become melanomas. Yes melanoma kills, really kills.
Now to the NY Times, it has a complaint about TSA X Ray machines. But a few years ago I wrote a long analysis of the X Ray scanners and melanoma. Yes it exists and X Rays are causitive. In fact look at my draft book on melanoma genomics.
The author states:
I have never walked through an airport body scanner — or, as I think of it, “the cancer machine.” In the years since these radiation chambers began appearing in airports across the United States, I have developed a variety of tricks to avoid submitting myself to them.
Good for the author. He continues:
The T.S.A. assures us that neither the X-ray scanners nor the millimeter wave machines pose a heath risk. But frankly I’d prefer to avoid being irradiated, even a little bit.
T.S.A. officers seem to take it personally when I opt out. They sigh, they roll their eyes, they snort derisively. I always have the impression that, at some point in their training, they have been told that passengers who opt out are foolish and selfish, because that is how I tend to be treated — with disgust.
Yes I have opted out. You see I have a predisposition. Then again it is the Government. Even more so it is TSA, one of the most troglodyte like elements of the current administrations task force of enforcers. Their science is not only defective but is harming Americans by the millions. Remember that it the Beltway Bandit firms who write the reports. These are the companies who hire the less than qualified and then produce expensive reports justifying Government attacks on it own citizens.
Now let me tell you how I really feel......
The TSA X Ray program is a major cause of latent cancers. It irradiates American citizens by the millions and does so with no understanding of the results. Perhaps that is why I drive everywhere. Or get the Fascist strip search. I really like Putin, at least he knows how to run a country!
Now to the NY Times, it has a complaint about TSA X Ray machines. But a few years ago I wrote a long analysis of the X Ray scanners and melanoma. Yes it exists and X Rays are causitive. In fact look at my draft book on melanoma genomics.
The author states:
I have never walked through an airport body scanner — or, as I think of it, “the cancer machine.” In the years since these radiation chambers began appearing in airports across the United States, I have developed a variety of tricks to avoid submitting myself to them.
Good for the author. He continues:
The T.S.A. assures us that neither the X-ray scanners nor the millimeter wave machines pose a heath risk. But frankly I’d prefer to avoid being irradiated, even a little bit.
T.S.A. officers seem to take it personally when I opt out. They sigh, they roll their eyes, they snort derisively. I always have the impression that, at some point in their training, they have been told that passengers who opt out are foolish and selfish, because that is how I tend to be treated — with disgust.
Yes I have opted out. You see I have a predisposition. Then again it is the Government. Even more so it is TSA, one of the most troglodyte like elements of the current administrations task force of enforcers. Their science is not only defective but is harming Americans by the millions. Remember that it the Beltway Bandit firms who write the reports. These are the companies who hire the less than qualified and then produce expensive reports justifying Government attacks on it own citizens.
Now let me tell you how I really feel......
The TSA X Ray program is a major cause of latent cancers. It irradiates American citizens by the millions and does so with no understanding of the results. Perhaps that is why I drive everywhere. Or get the Fascist strip search. I really like Putin, at least he knows how to run a country!
Treating the Patient vs Following the Book
There is an interesting article in JAMA comparing Evidenced (Guideline) Based Medicine versus Personalized Medicine. This is critical since the ACA fosters on physicians the EBM approach and may ultimately penalize them for deviating therefrom. After all it is the Government.
For EBM the authors clearly state:
Evidence-based guidelines are generated based on the body of clinical data available for a particular question. The highest level of evidence assigned in a guideline is based on multiple randomized controlled clinical trials. In general, randomized clinical trials have specific inclusion and exclusion criteria designed to represent a population broad enough and sufficiently enriched to attain a requisite number of end points and demonstrate a statistically and clinically significant difference in outcome. Subgroup analyses (both those that are prespecified and other post hoc analyses) are often performed to identify characteristics within the study population that are associated with greater benefit from the intervention, with no benefit, or even with harm. Yet these analyses are accompanied by warnings that findings should be cautiously interpreted.
The classic example is the prostate cancer tests and the breast cancer monitoring. The problem is always the issue of the subgroups and the users understanding of these limitations. The authors specifically state:
Indeed, there is well-deserved skepticism regarding the utility and accuracy of subgroup analysis from clinical trials, and these analyses are therefore generally not used in the formulation of guidelines. Patients (including those enrolled in trials) have multiple characteristics, each of which may influence the behavior and significance of other characteristics. Analysis of a subgroup showing that a single characteristic influences outcome is of limited clinical significance unless multiple variables that may modify the importance of the single variable are considered. However, if well-conducted analyses from multiple sources demonstrate concordant findings, perhaps these subgroup analyses should be considered when guidelines are constructed and revised, given the impracticality of performing randomized clinical trials to answer the question of appropriateness for every possible subgroup.
For example it is well known that prostate cancer is not the same in all men. Most is indolent and some highly aggressive. But how does one tell the difference. The "Guidelines" fail in all manners of reasonableness in addressing this.
Now the authors characterize Personalize Medicine as follows:
The President's Council of Advisors on Science and Technology noted that personalized medicine “refers to the tailoring of medical treatment to the individual characteristics of each patient. It does not literally mean the creation of drugs or medical devices that are unique to a patient, but rather the ability to classify individuals into subpopulations that differ in their susceptibility to a particular disease or their response to a specific treatment. Preventive or therapeutic interventions can then be concentrated on those who will benefit, sparing expense and side effects for those who will not.
Let us avoid any insight from the current president and his minions, for personalized medicine does really mean tailoring treatment down to the therapeutic level. For example, if we have a metastatic melanoma, then by understanding the receptor, ligand, pathway, and other elements broken in the mets we can develop not only a patient specific but CELL specific therapeutic. That is the future, and it is a future than can be achieved with what we have at hand today.
The authors conclude:
The conflict between guideline-based medicine and personalized medicine predominantly occurs when considering withholding a therapy that is recommended or supported by the guidelines but that may not be beneficial for an individual patient.
If a subpopulation may not benefit from the therapy, it is important to identify the subpopulation and verify this finding in an appropriate clinical trial. This presents a genuine opportunity to deliver better health care at lower costs by withholding the intervention. Cultivating a health care culture poised to explore these opportunities is critical.
This will entail more active participation from a range of stakeholders, including physicians who will need to embrace equipoise when the data support this position; insurers (including the Centers for Medicare & Medicaid Services) who have traditionally not been involved in the design, funding, and conduct of clinical trials; regulators who will need to develop policies to enable and support this type of patient-centered research; and health care organizations and quality-measurement groups who will need to develop more nuanced approaches to assessing quality of care and processes that monitor guideline implementation.
The key observation here is that as most physicians know all patients are different. Some benefit some do not. The reasons why are still often clouded in our ignorance. Will we allow our ignorance to dominate our treatments? Perhaps under ACA we not only will but must. Reimbursement may be the least of the problems. Failure to follow Guidelines, the "Rules", may very well place the physician at jeopardy. If the IRS has its say then perhaps failure to follow any Guideline may result in criminal charges, audits, and the like. This may be making physicians rule followers rather than diseases solvers.
For EBM the authors clearly state:
Evidence-based guidelines are generated based on the body of clinical data available for a particular question. The highest level of evidence assigned in a guideline is based on multiple randomized controlled clinical trials. In general, randomized clinical trials have specific inclusion and exclusion criteria designed to represent a population broad enough and sufficiently enriched to attain a requisite number of end points and demonstrate a statistically and clinically significant difference in outcome. Subgroup analyses (both those that are prespecified and other post hoc analyses) are often performed to identify characteristics within the study population that are associated with greater benefit from the intervention, with no benefit, or even with harm. Yet these analyses are accompanied by warnings that findings should be cautiously interpreted.
The classic example is the prostate cancer tests and the breast cancer monitoring. The problem is always the issue of the subgroups and the users understanding of these limitations. The authors specifically state:
Indeed, there is well-deserved skepticism regarding the utility and accuracy of subgroup analysis from clinical trials, and these analyses are therefore generally not used in the formulation of guidelines. Patients (including those enrolled in trials) have multiple characteristics, each of which may influence the behavior and significance of other characteristics. Analysis of a subgroup showing that a single characteristic influences outcome is of limited clinical significance unless multiple variables that may modify the importance of the single variable are considered. However, if well-conducted analyses from multiple sources demonstrate concordant findings, perhaps these subgroup analyses should be considered when guidelines are constructed and revised, given the impracticality of performing randomized clinical trials to answer the question of appropriateness for every possible subgroup.
For example it is well known that prostate cancer is not the same in all men. Most is indolent and some highly aggressive. But how does one tell the difference. The "Guidelines" fail in all manners of reasonableness in addressing this.
Now the authors characterize Personalize Medicine as follows:
The President's Council of Advisors on Science and Technology noted that personalized medicine “refers to the tailoring of medical treatment to the individual characteristics of each patient. It does not literally mean the creation of drugs or medical devices that are unique to a patient, but rather the ability to classify individuals into subpopulations that differ in their susceptibility to a particular disease or their response to a specific treatment. Preventive or therapeutic interventions can then be concentrated on those who will benefit, sparing expense and side effects for those who will not.
Let us avoid any insight from the current president and his minions, for personalized medicine does really mean tailoring treatment down to the therapeutic level. For example, if we have a metastatic melanoma, then by understanding the receptor, ligand, pathway, and other elements broken in the mets we can develop not only a patient specific but CELL specific therapeutic. That is the future, and it is a future than can be achieved with what we have at hand today.
The authors conclude:
The conflict between guideline-based medicine and personalized medicine predominantly occurs when considering withholding a therapy that is recommended or supported by the guidelines but that may not be beneficial for an individual patient.
If a subpopulation may not benefit from the therapy, it is important to identify the subpopulation and verify this finding in an appropriate clinical trial. This presents a genuine opportunity to deliver better health care at lower costs by withholding the intervention. Cultivating a health care culture poised to explore these opportunities is critical.
This will entail more active participation from a range of stakeholders, including physicians who will need to embrace equipoise when the data support this position; insurers (including the Centers for Medicare & Medicaid Services) who have traditionally not been involved in the design, funding, and conduct of clinical trials; regulators who will need to develop policies to enable and support this type of patient-centered research; and health care organizations and quality-measurement groups who will need to develop more nuanced approaches to assessing quality of care and processes that monitor guideline implementation.
The key observation here is that as most physicians know all patients are different. Some benefit some do not. The reasons why are still often clouded in our ignorance. Will we allow our ignorance to dominate our treatments? Perhaps under ACA we not only will but must. Reimbursement may be the least of the problems. Failure to follow Guidelines, the "Rules", may very well place the physician at jeopardy. If the IRS has its say then perhaps failure to follow any Guideline may result in criminal charges, audits, and the like. This may be making physicians rule followers rather than diseases solvers.


Sunday, May 26, 2013
Is There a Genetics of Human Obesity?
There seems to be a persistent search for the genetic basis of human obesity. Again the basic fact of life is that 3500 kcal equal one pound of body weight. So if you burn 2000 kcal per day and consume 2350 per day you gain one pound in ten days. That is a simple law. Genes or no genes.
Now in a recent NYAS paper the authors search for the elusive cause, neglecting the apparent truth. They state:
Although a number of significant findings have been made, it appears that very little of the apparent heritability of body mass index has actually been explained to date. New approaches for data analyses and advances in technology will be required to uncover the elusive missing heritability, and to aid in the identification of the key causative genetic underpinnings of obesity.
In the past two decades, family studies and animal models have helped us to identify many genetic
events associated with obesity. Subsequently, GWAS have driven the transition from primarily studying monogenic traits to ones of a more polygenic nature. GWAS have also revolutionized the genomics of obesity field, in that it offers an unbiased approach to uncover novel common genetic variants contributing to the pathogenesis of obesity.
Despite these great advances, the combined results of linkage, candidate gene, and GWAS approaches have explained very little of the variance in BMI, suggesting that there are still many genetic findings to be made, most likely being rarer variants exhibiting small effects. In order to fully characterize this missing heritability, larger and larger sample sizes are going to be required to improve statistical power. New technologies, such as next generation sequencing, will help us identify these elusive obesity-associated variants, particularly as the price of these techniques continues to drop.
In addition, most of variants that capture the association with obesity from current GWAS are not themselves causative. As such, how to move from association to causality remains a big challenge for common complex diseases like obesity. Therefore, we need to develop new approaches for analysis to characterize the true causative genes and perform functional studies to determine their roles in obesity. Once at least some of these challenges have been mastered, we will have a clearer picture of the genomics of obesity. This, in turn,will help us produce more efficacious therapies and will guide us on the path to personalized medicine.
Somehow the basic element of obesity is missed. How does a gene make you eat more? Perhaps you have a lower BMR but the equation holds for almost all humans. Why waste time on genes. One must tell fat people they are fat because they fail to control intake, it is their fault.
Now in a recent NYAS paper the authors search for the elusive cause, neglecting the apparent truth. They state:
Although a number of significant findings have been made, it appears that very little of the apparent heritability of body mass index has actually been explained to date. New approaches for data analyses and advances in technology will be required to uncover the elusive missing heritability, and to aid in the identification of the key causative genetic underpinnings of obesity.
In the past two decades, family studies and animal models have helped us to identify many genetic
events associated with obesity. Subsequently, GWAS have driven the transition from primarily studying monogenic traits to ones of a more polygenic nature. GWAS have also revolutionized the genomics of obesity field, in that it offers an unbiased approach to uncover novel common genetic variants contributing to the pathogenesis of obesity.
Despite these great advances, the combined results of linkage, candidate gene, and GWAS approaches have explained very little of the variance in BMI, suggesting that there are still many genetic findings to be made, most likely being rarer variants exhibiting small effects. In order to fully characterize this missing heritability, larger and larger sample sizes are going to be required to improve statistical power. New technologies, such as next generation sequencing, will help us identify these elusive obesity-associated variants, particularly as the price of these techniques continues to drop.
In addition, most of variants that capture the association with obesity from current GWAS are not themselves causative. As such, how to move from association to causality remains a big challenge for common complex diseases like obesity. Therefore, we need to develop new approaches for analysis to characterize the true causative genes and perform functional studies to determine their roles in obesity. Once at least some of these challenges have been mastered, we will have a clearer picture of the genomics of obesity. This, in turn,will help us produce more efficacious therapies and will guide us on the path to personalized medicine.
Somehow the basic element of obesity is missed. How does a gene make you eat more? Perhaps you have a lower BMR but the equation holds for almost all humans. Why waste time on genes. One must tell fat people they are fat because they fail to control intake, it is their fault.
Travel and Tourists
The NY Times has a small piece on travel. As the author states:
But not long ago, on a journey through India, I began to see things a little differently. For two weeks, I had been fairly battered by the daily chaos of budget travel. Then, on my last night in Kolkata, I met up with some particularly affluent friends who had spent their vacation escorted by private staff from one security-gated refuge to the next, and who were staying in a palatial five-star hotel on the outskirts of the city. In their cocoon of opulence, they quizzed me about my comical but vivid excursions, which had left me both exhausted and exhilarated. I began to realize that they suffered their own form of travel envy. The sense of control money provided them had also served to deaden their experience.
The economic gulf between travelers is part of a great tradition. Since the birth of leisure travel, aristocrats have been devising creative ways to isolate themselves from hoi polloi.
Now I have traveled a great deal. I have started companies in some twenty countries and can manage about six languages. Perhaps that is the key.
I am reminded of my first trip to Russia as a tourist. It was 1995 if I recall and we went via a FinnAir tour, but unlike the rest of the Americans we went local. We stayed in the InTourist hotel, the old KGB managed facility, where Russian was required, took local cabs to meet our rich American fellow travelers at the Europa.
When we went to the Ballet we had a ticket for $0.75 each since I dressed in a local style and spoke in Russian, the other Americans paid $75 from the Concierge. My tickets were from the local box office. We hired a local Russian driver, a fried of our InTourist guide, but since we spoke Russian, and dressed not as Americans, we were shown St Petersburg the way only a Russian would be. Later my Russian partners were always amazed that I would rather take the metro in Moscow than a limo.
In Thailand I managed to disappear into the countryside, with a few words of Thai, and in Italy my limited Sicilian allowed me to get places no American would be allowed.
After 9/11 I was stranded in France, and off to Normandy, where my French was accepted and the people were wonderful. We lived in Bayeux for the duration. We did not wear shirts festooned with logos, no sneakers, always had jackets, shirts, real shoes. A few words of the language and we saw the soul of the people.
I remember my days as a US corporate executive, the limos and managed tours, one saw nothing, one walked away from the opportunity with nothing.
I remember sitting at bars, speaking a little Russian and Greek, learning more of the world than any CIA analyst in Langley. Even more so, more than any Secretary of State.
Why does one travel, to escape, to learn, to mingle? Or perhaps if one has excess of funds just to show off. Yet than one can do that with less exercise.
But not long ago, on a journey through India, I began to see things a little differently. For two weeks, I had been fairly battered by the daily chaos of budget travel. Then, on my last night in Kolkata, I met up with some particularly affluent friends who had spent their vacation escorted by private staff from one security-gated refuge to the next, and who were staying in a palatial five-star hotel on the outskirts of the city. In their cocoon of opulence, they quizzed me about my comical but vivid excursions, which had left me both exhausted and exhilarated. I began to realize that they suffered their own form of travel envy. The sense of control money provided them had also served to deaden their experience.
The economic gulf between travelers is part of a great tradition. Since the birth of leisure travel, aristocrats have been devising creative ways to isolate themselves from hoi polloi.
Now I have traveled a great deal. I have started companies in some twenty countries and can manage about six languages. Perhaps that is the key.
I am reminded of my first trip to Russia as a tourist. It was 1995 if I recall and we went via a FinnAir tour, but unlike the rest of the Americans we went local. We stayed in the InTourist hotel, the old KGB managed facility, where Russian was required, took local cabs to meet our rich American fellow travelers at the Europa.
When we went to the Ballet we had a ticket for $0.75 each since I dressed in a local style and spoke in Russian, the other Americans paid $75 from the Concierge. My tickets were from the local box office. We hired a local Russian driver, a fried of our InTourist guide, but since we spoke Russian, and dressed not as Americans, we were shown St Petersburg the way only a Russian would be. Later my Russian partners were always amazed that I would rather take the metro in Moscow than a limo.
In Thailand I managed to disappear into the countryside, with a few words of Thai, and in Italy my limited Sicilian allowed me to get places no American would be allowed.
After 9/11 I was stranded in France, and off to Normandy, where my French was accepted and the people were wonderful. We lived in Bayeux for the duration. We did not wear shirts festooned with logos, no sneakers, always had jackets, shirts, real shoes. A few words of the language and we saw the soul of the people.
I remember my days as a US corporate executive, the limos and managed tours, one saw nothing, one walked away from the opportunity with nothing.
I remember sitting at bars, speaking a little Russian and Greek, learning more of the world than any CIA analyst in Langley. Even more so, more than any Secretary of State.
Why does one travel, to escape, to learn, to mingle? Or perhaps if one has excess of funds just to show off. Yet than one can do that with less exercise.
Tuesday, May 21, 2013
Treasury Spreads and Yield Curves



Sooner than later the FED will let go and these will increase substantially. When is always the question.
Sunday, May 19, 2013
How Dumb Do They Think We Are?

We are now at about 2.5% annual growth rate The CBO has it going to almost 6.5% in just a few years, by 2016. Who in their right mind believes that! Then it flat lines at 4.25%. What is the basis for that number? Try and read the report. If we cannot get true and reliable numbers with some basis, then why waste the time and money.
Things are really getting worse by the minute.
Thursday, May 16, 2013
MER, Melanoma and Inhibitors
The focus on pathways, receptors, ligands and promoters as
control elements for cancer has seen a great deal of development in the past
decade. One key approach is the development and identification of inhibitors,
molecules which can block an over excited pathway. We examine here a specific
recent such example as relates to melanoma. It is already well known that BRAF
suppression is an effective approach albeit often of limited duration. The
development of inhibitors for a selection of evolving pathway aberrations will
most likely be the way to turn a deadly disease into a chronic but manageable
problem, assuming that one can get permission to use such molecules, a process
which not is costly and lengthy.
We use a recent paper by Schlegel et al and use MER as a prototypical
example of pathway control via inhibitor blockage.
MER is a tyrosine kinase (“TK”) receptor (“TKR”)[1].
As Marks et al state there are 85 members in the TK family and 58 of these are
receptors. The receptors are divided into various families based upon their
structures and one family contains Axl, Sky and MER, also known as the TAM
family[2].
This family, as we shall see, has immunoglobulin like regions on the outside of
the cell surface and kinase domains on the inner surface. The family also has a
dual fibronectin III-like domain on the outside just below the immunoglobulin
domains, of which there are two.
Generally the receptors are activated by ligands which in
turn result in the phosphorlyation of the kinase region and associated area and
then commence the activation of the related pathways. Now these pathways are
the ones that result in proliferation and loss of localization and thus result
ultimately in metastasis.
We use this example for two reasons: (i) it is a good
example to demonstrate the activation of pathways and metastatic growth; (ii)
it also is a good example of how inhibitors can function on receptors and thus
can inhibit metastatic growth.
As Schlegel et al state:
Receptor tyrosine kinases (RTKs) are frequently
ectopically expressed, overexpressed, or hyperactivated in tumor cells and are therefore
attractive targets for cancer therapy. C-MER proto-oncogene tyrosine kinase
(MERTK), a member of the TAM (TYRO, AXL, MERTK) family of RTKs, has been
characterized as a therapeutic target in hematopoietic malignancies and several
solid tumors including lung, prostate, and brain
There is a subtle question posed but not answered here. Is
it over-expression, and if so by what ligand, or is it an excess production of
MER and thus an over-expression. What is the status of the benign cell, and is
this the dominant pathway. Clearly by having too active or too many MER
receptors, actually any TAM like receptor will do, leads to proliferation. This
goal of blocking the receptor so that it does not start the process is a valid
approach.
The authors clearly state:
Stimulation of melanoma cells with the MERTK ligand GAS6
resulted in the activation of several downstream signaling pathways including MAPK/ERK,
PI3K/AKT, and JAK/STAT. MERTK inhibition via shRNA reduced MERTK-mediated
downstream signaling, reduced colony formation by up to 59%, and diminished
tumor volume by 60% in a human melanoma murine xenograft model.
Namely we have a ligand, GAS6, which activates the MER
pathway. Is that ligand over expressed. On the other hand the molecule shRNA
reduced the activation.
They specifically state:
In addition, Sensi et al. found that melanoma cells often
secrete GAS6, a ligand of TAM receptors, indicating a mechanism of TAM
autocrine signaling in melanoma…. The mechanism of MERTK activation in melanoma
cells is not clear, but Sensi et al. have previously described melanoma cell
expression and secretion of GAS6, the common ligand for all members of the TAM family
of proteins, suggesting a method of autocrine and/or paracrine activation of MERTK.
Since expression of MERTK by melanoma cells increases during progression from
primary to metastatic melanoma, it would be interesting to determine whether
corresponding increases in GAS6 levels occur in serum from patients with
metastatic melanoma, implicating serum GAS6 levels as a potential early marker
of melanoma progression, as in other cancers.
Thus possibly inhibiting GAS6 may be profitable as well[3].
However the focus here is receptor inhibition.
1.1
MER and Melanoma
Let us consider a recent development in understanding MER
and melanoma. We return to the recent paper by Schlegel et al where the author’s
state:
C-MER proto-oncogene tyrosine kinase (MERTK) is a
receptor tyrosine kinase with oncogenic properties that is often overexpressed
or activated in various malignancies. Using both protein immunohistochemistry
and microarray analyses, we demonstrate that MERTK expression correlates with
disease progression.
MERTK expression was highest in metastatic melanomas,
followed by primary melanomas, while the lowest expression was observed in
nevi. Additionally, over half of melanoma cell lines overexpressed MERTK
compared with normal human melanocytes; however, overexpression did not
correlate with mutations in BRAF or RAS.
Stimulation of melanoma cells with the MERTK ligand GAS6
resulted in the activation of several downstream signaling pathways including
MAPK/ERK, PI3K/AKT, and JAK/STAT. MERTK inhibition via shRNA reduced
MERTK-mediated downstream signaling, reduced colony formation by up to 59%, and
diminished tumor volume by 60% in a human melanoma murine xenograft model.
Treatment of melanoma cells with UNC1062, a novel
MERTK-selective small-molecule tyrosine kinase inhibitor, reduced activation of
MERTK-mediated downstream signaling, induced apoptosis in culture, reduced
colony formation in soft agar, and inhibited invasion of melanoma cells. This
work establishes MERTK as a therapeutic target in melanoma and provides a
rationale for the continued development of MERTK-targeted therapies.
Thus, like to work that led to BRAF V600 inhibitors, we see
MER TK is another interesting target. The authors also provide an inhibitor
molecule as well.
1.2
Tyrosine Kinases and MER
Tyrosine Kinases receptors have received a great deal of
attention especially in the area of cancer metastasis and in cancer control.
They are as Verma et al state:
Receptor tyrosine kinases (RTK) are a large family of transmembrane
proteins exhibiting great diversity in their extracellular regions, although
sharing in common a highly conserved intracellular tyrosine kinase domain. They
function as sensors for extracellular ligands, the binding of which triggers
receptor dimerization and activation of the receptor’s kinase activity. This
activation leads to the recruitment, phosphorylation, and activation of
multiple downstream signaling proteins, which ultimately change the physiology
of the cell. RTKs regulate cellular processes, including survival, growth,
differentiation, adhesion, proliferation, and motility. Fifty-eight known RTKs
in the human genome are classified into 20 families by amino acid sequence
identity within the kinase domain and structural similarities within their extracellular
regions.
There are many such tyrosine kinase receptors. One class is
the TAM family and as Verma et al state:
One subfamily is referred to as the TAM family,
identified in 1991, comprising Tyro-3 (also called Sky), Axl, and Mer. The TAM
receptors are characterized by a combination of 2 immunoglobin-like domains and
dual fibronectin type III repeats in the extracellular region and a cytoplasmic
kinase domain. The primary ligand for TAM receptors is growth arrest-specific 6
(Gas 6), a fairly large (75 kDa) vitamin K–dependent protein known to activate
downstream signaling
We depict a simple structure below containing the elements
specified above.
Let us consider a simple development of MER controlled
pathways. The Figure below shows two separate and un-activated MERTK molecules
with the immunoglobulin terminals on the outside and the kinase areas on the
inside.

Now along comes a GAS6 ligand, and it attaches to and
connects the MERTK molecules at the immunoglobulin ends and this activates the
kinase tails inside the cell.

Once activated the kinase ends commence pathway activation
via the phosphorylation process. The pathways are depicted below.

It is the activation of these pathways by the excess GAS6
production or the excess MERTK production or both that results in excess
proliferation and metastasis.
As Verma et al relate about the pathway:
Studies using chimeric Mer receptors expressed in NIH3T3
fibroblasts linked downstream signaling pathways, such as PI3K, phospholipase
C-g (PLCg), and ERK, to Mer activation. Gas 6–dependent activation of Mer stimulates
phosphorylation of ERK1/2, leading to cellular transformation and increased
proliferation and DNA synthesis.
The ultimate downstream targets of the pathway differ
according to cell type and tissue microenvironment. In leukemia cells,
ligand-dependent activation of EGF receptor (EGFR)–Mer chimeric receptor
stimulates phosphorylation of Akt, ERK 1/2, and p38 mitogenactivated protein
kinases (MAPK), which results in decreased apoptosis but no change in
proliferation (30). Expression of CD8-Mer chimera in pro-B cells results in
transcriptional activation of NF-kB via PI3K/Akt.
Additional activation of p38/MAPK and meiosis-specific serine/threonine
protein kinase 1 (MEK1) occurs via CD8-Mer, leading to protection from
apoptosis. Some atypical signaling pathways involved in cell survival have been
studied as a link between Mer and the actin cytoskeleton via growth factor
receptor-bound protein 2 (Grb2), Shc, and Vav1. Downregulation of the proapoptotic
tumor suppressor WW domain-containing
We depict below how one can inhibit this process. We depict
an inhibitor molecule which binds to the sites as before but now does not
activate the TK pathways. The inhibitor must be stronger in affinity than the
GAS6 which most likely is still in ECM abundance.

Note above the RAs to RAF (especially BRAF) to MEK to MAPK
pathways flow. We have examined this in details elsewhere[4].
The implication is that by targeting the TK Receptor, one targets all elements
of the pathway. It should be noted however that the separate pathway elements
may be activated and over expressed via other factors such as epigenetic ones.
Thus the suggestions of Schlegel et al are of great merit but should be
balanced by understanding the epigenetic issues as well.
An example of a pathway and its control with BRAF
functionality is depicted below:

This simple explanation is also a paradigm for many other
such pathway activations and especially for those of the tyrosine kinase
verity.
1.3
MER and miRNA
There are other dimensions of interest here as well. In
cancers there unfortunately is not just a single point of failure. There often
are multiple. We show here just another example where MER and miRNA play an
interesting role. This is an essential point to make because all too often the
initial observers may all too often jump at a simple solution leaving behind a
complexity of other factors which take control.
Let us consider a miRNA control using MER. As Halberg et al
state:
Tumours require the establishment of vasculature for
their increasing nutrient, energy, and oxygen requirements as well as for
removal of metabolic waste. Cancer cells within a tumour generate such
pathologic vasculature by recruiting endothelial cells to the tumour site. This
is accomplished by secreting molecular factors, such as the well-known vascular
endothelial growth factor (VEGF, into the extracellular space.
VEGF binding to VEGF receptors on endothelial cells
results in the migration and recruitment of endothelial cells. In this way,
proteins expressed by cancer cells can regulate the cellular and structural
content of tumours—giving rise to continued tumour growth. Recent work has
revealed a major role for another class of genes— known as small non-coding
RNAs (microRNAs)—in the regulation of endothelial recruitment and tumour
angiogenesis.
One member of this family (miR-126) was recently found to
inhibit endothelial recruitment by suppressing a set of cancer genes that
activate endothelial migration. In this way, a non-coding RNA expressed by
cancer cells could shape the tumour and metastatic microenvironment.
This is thus depicted below from the Halberg paper. Here we
have two cells, the top cell is a cancer cell where miR126 is blocking IGBP2
and blocking the MERTK receptor which in turn would have blocked the entrance
of GAS6. But since miR126 has blocked the blocker, we have excess GAS6. Thus we
have a problem, namely the GAS6 “overproduction” is really a failure to block
resulting from the cancer cell miR126 production.

It is critical always therefore to look across all paths,
direct as well as epigenetic.
As Zhuang et al state:
Angiogenesis plays a crucial role during tumorigenesis
and much progress has been recently made in elucidating the role of VEGF and
other growth factors in the regulation of angiogenesis. Recently, microRNAs
(miRNAs) have been shown to modulate a variety of physiological and
pathological processes.
We identified a set of differentially expressed miRNAs in
microvascular endothelial cells co-cultured with tumour cells. Unexpectedly,
most miRNAs were derived from tumour cells, packaged into microvesicles (MVs), and
then directly delivered to endothelial cells.
Among these miRNAs, we focused on miR-9 due to the strong
morphological changes induced in cultured endothelial cells. We found that
exogenous miR-9 effectively reduced SOCS5 levels, leading to activated JAK-STAT
pathway. This signalling cascade promoted endothelial cell migration and tumour
angiogenesis.
Remarkably, administration of anti-miR-9 or JAK
inhibitors suppressed MV-induced cell migration in vitro and decreased tumour
burden in vivo. Collectively, these observations suggest that tumour-secreted
miRNAs participate in intercellular communication and function as a novel
pro-angiogenic mechanism.
1.4
Inhibitors
Inhibitors of pathways are being developed at a rapid rate.
Knowing the pathway and molecular structure of the receptors it is somewhat
readily possible to develop a strong inhibitor, a molecule that interferes with
the normal ligand.
Schlegel et al have developed and tested an inhibitor of the
MERTK receptor and it is shown below.

Schlegel et al characterize this molecule as follows:
A novel MERTK tyrosine kinase inhibitor, UNC1062,
inhibits MERTK mediated signaling, promotes apoptosis, and inhibits colony
formation in melanoma cells. While activating mutations in BRAF and NRAS occur
in melanoma at rates of 41% and 18%, respectively, lower mutation frequency or
gene amplifications in other signaling molecules, such as RTKs, can also
contribute to melanoma pathogenesis.
UNC1062 was
developed as a MERTK-selective tyrosine kinase inhibitor. Its structure is based
on a previously published pyrazolopyrimidine scaffold, and it has an improved
affinity and specificity profile compared with its parent compound, UNC569 .
UNC1062 potently inhibits MERTK kinase activity in vitro
and exhibits specificity within the TAM family. Treatment of HMCB and G361
cells with increasing concentrations of UNC1062 resulted in a potent
dose-dependent reduction in MERTK phosphorylation
In the work of Verma et al they present an interesting
collection of molecules which exhibit inhibitor characteristics (see their
Figure 2). This is an expansion of what Schlegel et al have presented.
Again from Schlegel et al we have:
MAPK/ERK and PI3K/AKT are 2 of the most frequently dysregulated
pathways in melanoma. These 2 pathways not only play a role in melanoma
development and progression, but are also involved in primary and secondary
resistance to BRAF inhibitors.
The observation that MERTK signals via both pathways, as
well as through others whose roles in melanoma biology are currently unclear
(e.g., STAT6), not only highlights the complex regulation of these pathways by
membrane receptors, such as MERTK, but may also provide a therapeutic
advantage, since targeting MERTK may disrupt signaling in multiple pathways.
These observations and the data presented here suggest
that MERTK-targeted therapies could potentially be considered for patients,
irrespective of BRAF and NRAS status and/or prior treatment with BRAF
inhibitors.
The latter observation is of possible significant merit. Namely
the MERTK targeting allows for alternative pathway blocking, namely doing so at
the source of pathway activation.
1.5
Observations
We conclude with some general and specific observations.
This work by Schlegel et al is of significant importance for reasons already
indicated.
1. MERTK presents an attractive target for metastatic
diseases.
To best summarize, we use the words directly from the paper.
Schlegel et al conclude:
We believe this work has led to several novel insights.
First, MERTK expression is significantly elevated in
distant metastatic tumors compared with primary melanomas.
Second, MERTK is overexpressed in approximately half of
melanoma cell lines, irrespective of BRAF and NRAS status, and is an active
receptor.
Third, targeting MERTK suppresses prosurvival pathways
such as STAT6, AKT, and ERK1/2.
Fourth, targeting MERTK suppresses colony-forming
potential and migration.
And fifth, targeting MERTK in vivo retards tumor growth
in a human melanoma xenograft model.
The finding that MERTK expression is highest in distant
metastatic melanomas compared with primary melanomas and the roles of MERTK in
colony formation, migration, and invasion suggest that MERTK plays a role in
the progression of primary melanomas and the development of distant metastases.
Similar to the observations in this report, the migratory
nature of glioblastoma cells could be reduced by MERTK inhibition with either
shRNA knockdown or a MERTK monoclonal antibody, suggesting that increased MERTK
expression may contribute to outgrowth of the metastatic tumor.
2. MER and other TAM receptors show significant impact across
broad areas of cancer activity.
Now Verma et al present an interesting summary table as show
below which recounts what cancer types are also upregulated TAM pathways. The
breath of such upregulation is significant. It also may present significant
opportunities for blockage molecules, namely inhibitors of the total pathway.

3. GAS6 Inhibition on MERTK by inhibitors is an
attractive approach to metastatic melanoma
Developing receptor inhibitors is a powerful approach to
controlling metastatic growth and proliferation.
As Verma et al state:
A potential ability of sAxl to serve as a natural
antagonist of Gas 6 could have clinical relevance. Similarly, the
membrane-bound Mer protein is cleaved in the extracellular domain via a
metalloproteinase (38). Further studies are needed to establish sAxl and sMer
as important biomarkers for correlation with disease stage and predicting
prognosis.
As Segal et al state:
A subset of genes, including the small monomeric GTPase
RABB33, the proto-oncogene MERTK, the glycopeptide hormone STC1, and the
neuropeptide GAL were shown to discriminate CCS/MSP from both STS and melanoma.
We further surveyed specific genes of interest and found
melanoma differentiation antigens TYRP1, TYRP2/DCT, and MART-1 to be expressed
at varying levels in the CCS/MSP specimens. PMEL17 was most consistently
expressed in all four tumors in a similar distribution to that of MITF.
Interestingly, SOX10, which induces MITF expression, was expressed in all
CCS/MSP and most melanoma specimens
Thus it appears that this approach and ones like it are
useful for thorough examination as attractive and effective means of metastatic
control and management.
However there is still a long way from this point to
approved therapeutics.
1.6
References
1. Halberg, N., et al, microRNA regulation of cancer–endothelial
interactions: vesicular microRNAs on the move, The EMBO Journal (2012) 31,
3509–3510.
2. Marks, F., et al, Cellular Signal Processing, Garland (NY) 2009.
3. Park, H. et al, The TAM-family receptor Mer mediates production
of HGF through the RhoA-dependent pathway in response to apoptotic cells, Molecular
Biology of the Cell, Volume 23 August 15, 2012.
4. Schlegel, J., et al, MERTK receptor tyrosine kinase is a
therapeutic target in melanoma, The Journal of Clinical Investigation, http://www.jci.org,
Volume 123, Number 5, May 2013 p.2257.
5. Segal, N., et al, Classification of Clear-Cell Sarcoma as a
Subtype of Melanoma by Genomic Profiling, J Clin Oncol 21:1775-1781, 2003.
6. Verma, A., et al, Targeting Axl and Mer Kinases in Cancer, Mol
Cancer Ther 2011; 10: 1763-1773.
7. Zhuang, G., et al, Tumour-secreted miR-9 promotes endothelial
cell migration and angiogenesis by activating the JAK-STAT pathway, The EMBO
Journal 31, 3513 - 3523 (6 July 2012) http://www.nature.com/emboj/journal/v31/n17/full/emboj2012183a.html
[1] From
NCBI we have (2q14.1): This gene is a member of the MER/AXL/TYRO3 receptor
kinase family and encodes a transmembrane protein with two fibronectin type-III
domains, two Ig-like C2-type (immunoglobulin-like) domains, and one tyrosine
kinase domain.
[2]
TAM, (TYRO, AXL, MERTK)
[3] As
NCBI states: This gene product is a gamma-carboxyglutamic acid
(Gla)-containing protein thought to be involved in the stimulation of cell
proliferation, and may play a role in thrombosis. Alternatively spliced
transcript variants encoding different isoforms have been found for this gene.
Located at 13q34. http://www.ncbi.nlm.nih.gov/gene/2621
[4]
See McGarty, Melanoma Genomics, DRAFT, 2013.
Wednesday, May 15, 2013
Power Lines and Leukemia
There has been an ongoing debate as to high power lines and cancer, especially leukemia amongst children. A recent study from France alleges a correlation, apparently without causation.
The study in BJC states:
The present study, free from any participation bias, supports the previous international findings of an increase in AL incidence close to VHV-HVOL. In order to investigate for a potential role of ELF-MF in the results, ELF-MF at the residences close to HVOL are to be estimated, using models based on the annual current loads and local characteristics of the lines.
Increased odds ratios (ORs) were observed for AL occurrence and living within 50m of a VHV-HVOL (OR¼1.7 (0.9–3.6)). In contrast, there was no association with living beyond that distance from a VHV-HVOL or within 50m of a HV-HVOL....
In conclusion, the present study has generated additional findings, based on a recent nationwide unselected population-based study, that support the hypothesis that living o50m from a 225 or 400 kV HVOL may be associated with an increased incidence of childhood AL. No increase in risk was observed further from those lines and no increase in childhood AL risk was detected within 50m of the 63–150 kV HVOL. Model-based estimates of ELF-MF exposures will be used to investigate for potential involvement of ELF-MF in the observed association.
It does leave one wondering what the cause is. One is always concerned about correlation and causation. Especially here.
The study in BJC states:
The present study, free from any participation bias, supports the previous international findings of an increase in AL incidence close to VHV-HVOL. In order to investigate for a potential role of ELF-MF in the results, ELF-MF at the residences close to HVOL are to be estimated, using models based on the annual current loads and local characteristics of the lines.
Increased odds ratios (ORs) were observed for AL occurrence and living within 50m of a VHV-HVOL (OR¼1.7 (0.9–3.6)). In contrast, there was no association with living beyond that distance from a VHV-HVOL or within 50m of a HV-HVOL....
In conclusion, the present study has generated additional findings, based on a recent nationwide unselected population-based study, that support the hypothesis that living o50m from a 225 or 400 kV HVOL may be associated with an increased incidence of childhood AL. No increase in risk was observed further from those lines and no increase in childhood AL risk was detected within 50m of the 63–150 kV HVOL. Model-based estimates of ELF-MF exposures will be used to investigate for potential involvement of ELF-MF in the observed association.
It does leave one wondering what the cause is. One is always concerned about correlation and causation. Especially here.
Monday, May 13, 2013
Bowman v Monsanto

Held: Patent exhaustion does not permit a farmer to reproduce patented seeds through planting and harvesting without the patent holder’s permission.
(a) Under the patent exhaustion doctrine, “the initial authorized sale of a patented article terminates all patent rights to that item,” Quanta Computer, Inc. v. LG Electronics, Inc., 553 U. S. 617, 625, and confers on the purchaser, or any subsequent owner, “the right to use [or] sell” the thing as he sees fit, United States v. Univis Lens Co., 316 U. S. 241, 249–250. However, the doctrine restricts the patentee’s rights only as to the “particular article” sold, id., at 251; it leaves untouched the patentee’s ability to prevent a buyer from making new copies of the patented item. By planting and harvesting Monsanto’spatented seeds, Bowman made additional copies of Monsanto’s patented invention, and his conduct thus falls outside the protections of patent exhaustion. Were this otherwise, Monsanto’s patent would provide scant benefit. After Monsanto sold its first seed, other seed companies could produce the patented seed to compete with Monsanto, and farmers would need to buy seed only once.
(b) Bowman argues that exhaustion should apply here because he is using seeds in the normal way farmers do, and thus allowing Monsanto to interfere with that use would create an impermissible exception to the exhaustion doctrine for patented seeds. But it is really Bowman who is asking for an exception to the well-settled rule that exhaustion does not extend to the right to make new copies of the patented item. If Bowman was granted that exception, patents on seeds would retain little value. Further, applying the normal rule will allow farmers to make effective use of patented seeds. Bowman, who purchased seeds intended for consumption, stands in a peculiarly poor position to argue that he cannot make effective use of his soybeans. Bowman conceded that he knew of no other farmer who planted soybeans bought from a grain elevator. In the more ordinary case, when a farmer purchases Roundup Ready seed from Monsanto or an affiliate, he will be able to plant it in accordance with Monsanto’s license to make one crop.
The issue is simply that Monsanto developed a seed protecting itself against Roundup. However when corn is made from the seed, it is made by cross fertilizing naturally with plants like itself and others. Nature does the mixing. Bowman took the F1 seed, namely the first cross, and planted it. He then treated it with Roundup killing off the plants without the gene protection and selecting the good genes. The Court then says by this ruling that you may genetically engineer something, and under the old interpretation you protected the something you engineered, via vegetative propagation. But now it protects against cross propagation by fertilizing. This is a dramatic extension of plant patents. The old law allowed you to patent a purple flower. You then propagated it by cuttings and it was still yours. However if you used it to cross with another flower the resulting plants were yours. Perhaps no longer!
Does that mean I need the Patent basis for every plant I use, and for how many generations. This decision creates a real mess. It is a shame out Judges have no knowledge of Biology.
Subscribe to:
Posts (Atom)


