In a recent paper by Forloni et al the authors state:
Oncogenic mutations in BRAF and NRAS occur in 70% of
melanomas. In this study, we identify a specific microRNA, miR-146a, which is
highly upregulated by oncogenic BRAF and NRAS. Expression of miR-146a increases
the ability of human melanoma cells to proliferate in culture and form tumors
in mice, whereas knockdown of miR-146a has the opposite effects. We show these
oncogenic activities are due to miR-146a targeting the NUMB mRNA, a repressor
of Notch signaling.
The focus is now clearly on these secondary factors, namely
the micro RNAs and even methylation effects that are seen in many cancers. In
this case it of the excess production of a specific miRNA that in turn block an
mRNA and in turn allows upregulation of other pathways and in turn unregulated
cell proliferation.
As Garraway states in NEJM:
Finally, these findings invite speculation that adding
γ-secretase inhibitors to inhibitors of RAF and MEK might offer an attractive
therapeutic cocktail for assessment in future clinical trials of melanoma
treatment. Given the substantial toxicity of γ-secretase inhibitors, additional
preclinical studies of such combinations in melanoma cell lines and patient
derived xenograft models would be beneficial.
Such studies could clarify the generalizability of Notch
dependency in melanoma, the relevance (if any) of the pre miR146a G allele
versus the C allele for patient stratification, and the possible usefulness of
alternative dosing and scheduling schema to reduce toxicity. Overall, this
study provides a reminder that, despite numerous advances, we have only just
begun to dissect the rich interplay among noncoding RNAs, the biologic basis of
cancer, and potential therapeutic strategies.
Garraway sees this as a significant breakthrough and believe
it is a worthwhile pathway for new therapeutics. In this analysis we briefly
examine the interaction of Notch and miR-146a and how it can be understood in
the case of melanoma as a major factor of uncontrolled proliferation.
Proteolysis is the process of degradation of proteins in the
cell and the release of the energy contained therein for other purposes. The
Notch pathway process is a key part of the proteolysis effort[1]. The
Notch system is a proteolytic driven system used in signal transduction in
cells. Uncontrolled Notch pathways production can lead to uncontrolled cellular
growth.
Let us begin with a simplified but reflective description of
the Notch pathway. The Notch process starts with the two Notch ligands, which
are also called DSL proteins. One is external to the cell membrane and is the
other is internal. When they are broken, the intracellular part, called NICD
moves to the nucleus and binds with a protein CSL which becomes a putative
transcription factor. We demonstrate that below.

Recall, that a transcription factor is a protein or protein
complex that can turn on (activators) or turn off (repressors) the transcription
of genes[2].
In this case the transcription factor is an activator for MYC[3].
Transcription factors are frequently brought to bear to activate genes that
lead to uncontrolled growth.
Goss and Kahn have presented a review of the interaction of
Notch and Wnt and especially the function of excess Notch activation as a part
of cell proliferation in multiple cancers[4].
As they state Wnt and Notch act in concert in many cancers, prostate being one
which we have examined in some detail. In addition excess Notch activation
appears to effect a stem cell like behavior in these cells thus resembling the
cell types enable for proliferation as well as survival.
We now want to explore some of the impacts of Notch in stem cell
environments and in turn in the maturation of cells. We focus on a recent paper
by Katoh and Katoh. As Katoh and Katoh have written:
Notch signaling pathway is implicated in the maintenance
of self-renewal potential in stem cells, binary cell-fate determination in
progenitor cells, and induction of terminal differentiation in proliferating
cells. Notch-ligand binding to Notch receptors leads to the cleavage of Notch
receptors by metalloprotease and Á-secretase to induce nuclear translocation of
Notch intracellular domain (NICD). Nuclear complex, consisting of CSL (RBPSUH),
NICD, Mastermind (MAML), p300 and histone acetyltransferase (HAT), then induces
transcriptional activation of Notch target genes, such as HES1, HES5, HES7,
HEY1, HEY2 and HEYL. HES/HEY family members are bHLH-type transcriptional
repressors for tissue-specific transcription factors. Therefore, Notch
signaling activation in stem cells leads to the maintenance of self-renewal
potential.
Now Katoh and Katoh provide an activation path progression
as show below (as modified):

The above demonstrates the progress from an overactive Notch
cell which thus acts as a stem cell to more mature cell lines. The above also
demonstrates the location of proliferating cells in this schema, just after the
stem cell line progenitor.
Thus the activation of Notch leads to an extreme survival
capability in cells so activated. They continue with the following regarding
NUMB:
NUMB and NUMB-like (NUMBL), consisting of phosphotyrosine-binding
(PTB) domain and SH3-binding proline-rich region, are docking proteins
functioning as Notch signaling inhibitors. Here, we searched for the
TCF/LEF-binding site within NUMB and NUMBL promoters. Because two
TCF/LEF-binding sites were identified within human NUMB promoter, comparative
integromics analyses on NUMB orthologs were further performed.
Thus one way to over-activate Notch is to suppress NUMB.
NUMB is described by NCBI as follows[5]:
The protein encoded by this gene plays a role in the
determination of cell fates during development. The encoded protein, whose
degradation is induced in a proteasome-dependent manner by MDM2, is a
membrane-bound protein that has been shown to associate with EPS15, LNX1, and
NOTCH1.
In a similar manner NOTCH1 is described as follows[6]:
This gene encodes a member of the Notch family. Members
of this Type 1 transmembrane protein family share structural characteristics
including an extracellular domain consisting of multiple epidermal growth
factor-like (EGF) repeats, and an intracellular domain consisting of multiple,
different domain types. Notch family members play a role in a variety of
developmental processes by controlling cell fate decisions.
The Notch signaling network is an evolutionarily
conserved intercellular signaling pathway which regulates interactions between
physically adjacent cells. …Homologues of the notch-ligands have also been
identified in human, but precise interactions between these ligands and the
human notch homologues remain to be determined. This protein is cleaved in the
trans-Golgi network, and presented on the cell surface as a heterodimer. This
protein functions as a receptor for membrane bound ligands, and may play
multiple roles during development.
These are two powerful and interacting genes. NUMB
suppresses Notch1 and Notch1 when activated makes for cell proliferation and
survival.
There are now well over hundreds of micro RNAs, which a
small non-coding RNAs which result in the control of various pathways in cellular
signalling. Micro RNAs are often encoded in introns in mRNAs and some in in
non-coding RNAs. They generally control mRNA in terms of its stability,
degradation and/or translation. The micro RNAs can stop genes from being
expressed as proteins, even though the gene is present and provides a normal
mRNA. They are small, generally 22 base pairs in length.
As NCBI states[7]:
microRNAs (miRNAs) are short (20-24 nt) non-coding RNAs
that are involved in post-transcriptional regulation of gene expression in
multicellular organisms by affecting both the stability and translation of
mRNAs. miRNAs are transcribed by RNA polymerase II as part of capped and
polyadenylated primary transcripts (pri-miRNAs) that can be either
protein-coding or non-coding.
The primary transcript is cleaved by the Drosha
ribonuclease III enzyme to produce an approximately 70-nt stem-loop precursor
miRNA (pre-miRNA), which is further cleaved by the cytoplasmic Dicer
ribonuclease to generate the mature miRNA and antisense miRNA star (miRNA*)
products. The mature miRNA is incorporated into a RNA-induced silencing complex
(RISC), which recognizes target mRNAs through imperfect base pairing with the
miRNA and most commonly results in translational inhibition or destabilization
of the target mRNA.
As Rusca and
Monticelli state:
Initial evidences on the possible involvement of miR-
146a in cancer came from a study showing thatmiR-146a was upregulated in
papillary thyroid carcinoma (PTC) samples compared with unaffected thyroid
tissue. Interestingly, a set of five miRNAs, including miR-221, miR-222, and
miR- 146, was sufficient to distinguish unequivocally between PTC and normal
thyroid. Similarly to the observations performed in immunologic settings,
overexpression of miR- 146a/b in the highly metastatic human breast cancer cell
line MDA-MB-231 significantly downregulated expression of IRAK1 and TRAF6,
negatively regulating NF-κB activity. Functionally, this resulted in markedly
impaired invasion and migration capacity relative to control cells.
These findings implicated miR-146 not only as a negative
regulator of constitutive NF-κB activity in breast cancer cells, but also
suggested that modulating miR-146 levels might have therapeutic potential to
suppress breast cancer metastases. Along the same line, miR-146a was among the
miRNAs found upregulated in cervical cancer tissues compared to normal cervix.
When introduced into cell lines, miR- 146a promoted cell
proliferation. Although the molecular mechanism underlying such increased
proliferation remains to be investigated, these observations potentially
implicate miR-146a in cervical carcinogenesis. In another type of cancer, the
hormone-refractory prostate carcinoma (HRPC), miR-146a levels were diminished
compared to androgen-sensitive noncancerous epithelium. In this context, miR-
146a acted as a tumor suppressor, reducing levels of its target ROCK1, one of
the key kinases involved in HRPC transformation.
Accordingly, forced miR-146a expression reduced ROCK1
protein levels, cell proliferation, invasion, and metastasis to human bone
marrow endothelial cell monolayers. Similarly, miR-146a was lower in pancreatic
cancer cells compared with normal human pancreatic cells…
There is now increasing evidence to suggest that miR-146a
is involved in the regulation of the adaptive as well as innate immune
response, and that miR-146a can be an important player in regulating tumor
progression.
However, more work remains to be done to fully understand
its role and mechanism of action in normal and pathologic conditions, so that
expression of this miRNA can potentially be exploited as a new point of entry
for therapy. With the identification of a vast number of miRNAs each carrying a
long list of putative targets, the challenge is now to understand the details
of their biological functions.
Thus miR-146a has significant roles to play in controlling
cell behavior.
For example, miRNAs can inhibit the translation of mRNA into
a protein. We show this below. The small segment attaches to the mRNA and
blocks translation. This graphic is descriptive and does not contain full
details[8].

In the case being discussed, miR-146a binds to NUMB and
suppresses it. That in turn allows for an overexpression of Notch which in turn
can lead to an unstable system with feedback. We shall detail that a bit later.

We depict that process in some detail below. For the most
part all miRNAs appear to function in the same manner. There are well over a
thousand identified at this point and more than likely many more to be found.
The functions of most are not fully known.

Before continuing it is worth a quick review of normal and
abnormal behavior of miRNA. The normal process is shown below. This shows a
classic blocking of translation. The miRNA binds to the mRNA and inhibits
translation. The question is what makes the miRNA to do this? Namely what
forces the generation of the miRNA? Is it a random effect or is it part of a
planned process. We have shown that homeostasis is well defined in terms of a
balanced expression of RNA. Yet when we have an aberrant genetic element the
miRNA can express itself in deleterious ways.

Now we can examine a miRNA in the context of a cancerous
environment shown below. The diagram below shows miRNA blocking a tumor
suppressor gene. In a sense the example of the miR-146a is an example of this
type of miRNA operation. It blocks a protein which in turn blocks a protein
which leads to unbridled growth and survival, Notch.

Finally we show the example of miRNA in some explosive
expansion of itself thus blocking many tumor suppressor genes. This is a deadly
mode for miRNAs and can be found in many cancers[9].

The classic pathway dynamics we understand regarding proliferation
and survival is shown below. This is the BRAF and PI3K dynamics. We demonstrate
this below. This is a well-known and well understood pathway and is at the core
of the BRAF V600 therapeutic approach.

Now proliferation and survival require gene activation and
maintenance.
In this report, we demonstrate a critical role for
miR-146a in the initiation and progression of BRAF/ NRAS-positive melanomas,
... In addition, our results reveal a pharmacologically tractable pathway for
the treatment of melanoma. We identified miR-146a as the microRNA whose
expression was most upregulated by activated BRAF.
Upregulation of miR-146a by activated BRAF, as well as
activated NRAS, occurs through the MAPK signaling pathway. Accordingly, we find
that BRAF and NRAS mutant melanoma cell lines and short-term melanoma cultures
show higher levels of miR-146a compared to those that are wild type for these
genes.
A major function of the MAPK pathway is to activate
transcription by regulating the stability and expression of multiple
transcription factors primarily through direct phosphorylation.
We show that the MAPK pathway regulates the
phosphorylation of the transcription factor MYC, which in turn binds to the
promoter of miR-146a and stimulates its transcription. Notably, MYC has been
found to stimulate transcription of several other miRNAs. For example, MYC has
been shown to directly activate transcription of the oncogenic miR-17-92
cluster and thereby promote cell proliferation, survival, angiogenesis, and
metabolic reprogramming in a number of tumor cell lines.
miRNAs and components of miRNA biogenesis pathways such
as Dicer have been implicated in several aspects of melanocyte biology as well
as in melanoma initiation and progression.
We depict some of that process below:

Previous studies
have shown that miR-146a can function either as an oncogene or as a tumor
suppressor depending upon the cell type. For example, miR-146a has been shown to
function as an oncogene in a variety of human cancers including papillary
thyroid carcinoma (PTC), triple negative sporadic breast cancers and anaplastic
thyroid carcinoma. miRNAs function primarily by targeting mRNAs and either
promoting their degradation or blocking their translation.
Our analysis identified 20 potential targets of miR-146a,
including NUMB, which is a well-characterized Notch signaling inhibitor. It is
thought that NUMB negatively regulates NOTCH, potentially through a direct
protein–protein interaction that requires the phosphotyrosine-binding (PTB)
domain of NUMB and either the RAM23 region or the very C-terminal end of NOTCH.
We demonstrate some of these dynamics in the Figure below
(adapted from Galloway with modifications).
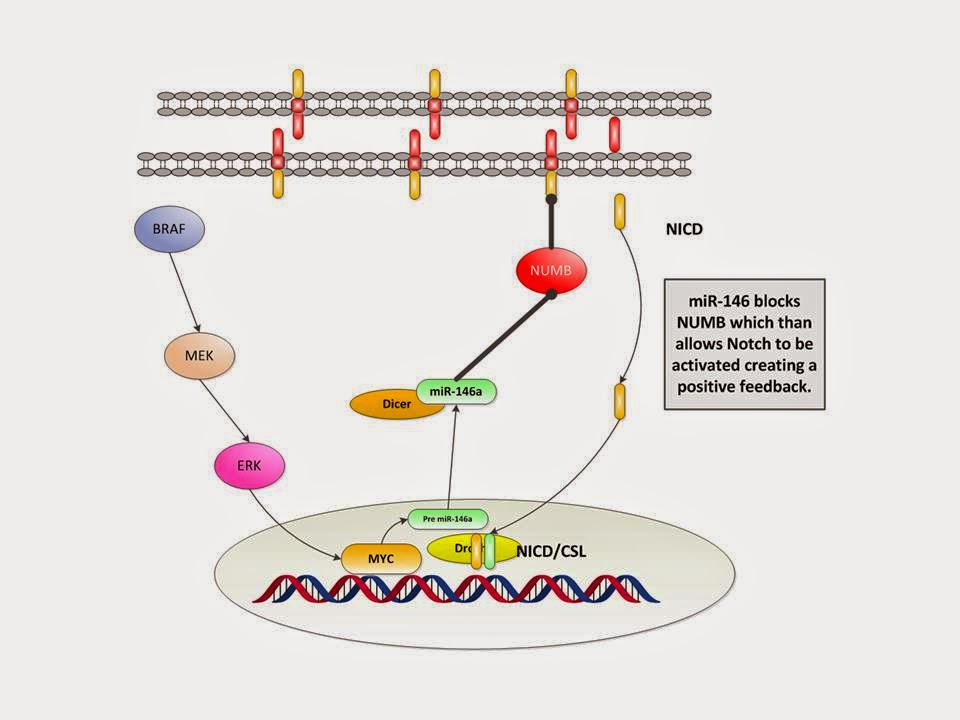
In simple terms:
1. BRAF activates MEK
2. and then ERK
3. which activates MYC
4. which activates pre miR-146a and
5. then via Drosha and Dicer makes miR-146a
6. which reactivates Notch by suppressing NUMB expression (we have
left that out for simplicity)
7. which then goes down to the transcription on the DNA resulting
in proliferation and stem like behavior.
This is an interesting and compelling mechanism for the
explanation of the aggressive melanoma expansion.
This is an interesting step in the understanding of melanoma
genomics. The role of micro RNAs is becoming clearer as time goes by and added
to that is the effect of such epigenetic factors as methylation and we now see
a much more complex field of play than a decade ago. The benefit is the
recognition of more targets of opportunity that can be had for potential
therapeutics. On the other hand the main concern is that the more that is
learned one may ask what else is there yet to grasp.
Thus what observations can we make here? Let us examine a
few:
1. Stem Cell Hypothesis. Here we have the elements of how a
stem cell functions with the activated Notch and blocked NUMB. Does this imply
that we have the re-emergence of stem cell like malignant cells activated in a
manner such as this. Namely the miRNAs allow for the reprogramming of some
modified form of totipotency.
2. Targeted Therapeutics: Galloway makes this observation We
know that BRAF inhibitors get us one step there but then we need MEK
inhibitors. Then what? Does an inhibitor for miR-146a take all the steps
necessary or does the cell go and find another back door way to function?
3. The Dynamics of the Processes are Not Well Understood.
One of the problems in understanding the impact of miRNAs and other pathway
elements is that there is a concern as to the number or concentration of
products. If miR-146a is to block NUMB then it should block all NUMB and in
turn activate all Notch. Yet it is a molecule by molecule process which seems
to be poorly understood. The paper by Choir et al and Nazarov et al present
some ideas on how to deal with such issues. However they are but first steps.
This is a critical factor to understand since the therapeutics depends on
blocking the necessary number of miR-146a molecules. To data there seems to be
limited data to assess this issue.
4. Initiation and Support: We know that V600 mutation of
BRAF is drivers for metastatic melanoma. However it is not clear what is the
driver ultimately for miR-146a, although it appears as we have suggested as a
sequella from the other mutations. Additional insight into the proliferation is
requires.
1.
Appasani, K., MicroRNAs,
Cambridge (New York) 2008.
2.
Broad Institute, https://www.broadinstitute.org/education/glossary
3.
Choi, M., et al, A dynamic
expression survey identifies transcription factors relevant in mouse digestive
tract development, Development 133, 4119-4129 (2006) doi:10.1242/dev.02537
4.
Forloni, M., et al,
miR-146a promotes the initiation and progression of melanoma by activating
Notch signaling, eLife 2014;3: e01460. DOI: 10.7554/eLife.01460
5.
Garraway, L., A Notch for
Noncoding RNA in Melanoma, NEJM, 370;20, May 15, 2014.
6.
Goss, K., M. Kahn,
Targeting the Wnt Pathway in Cancer, Springer (New York) 2011.
7.
Katoh, M., M., Katoh, NUMB
is a break of WNT - Notch signaling cycle, INTERNATIONAL JOURNAL OF MOLECULAR
MEDICINE 18: 517-521, 2006
8.
Lawrie, C., MicroRNAs in
Medicine, Wiley (Hoboken) 2014.
9.
Marks, F., et al, Cellular
Signal Processing, Garland 2008.
10.
McGarty, T., Melanoma
Genomics, DRAFT 2013.
11.
McGarty, T., Prostate
Cancer Genomics, DRAFT, 2013.
12.
Nazarov, P., et al,
Interplay of microRNAs, transcription factors and target genes: linking dynamic
expression changes to function, Nucleic Acids Research, 2013, Vol. 41, No. 5
2817–2831.
13.
Rusca, S., C. Monticelli,
MiR-146a in Immunity and Disease, Molecular Biology International, Volume 2011,
Article ID 437301, 7 pages
14.
Sangunitti, et al,
Probabilistic inference of transcription factor concentrations and
gene-specific regulatory activities, Bioinformatics, Vol. 22 no. 22 2006, pages
2775–2781
15.
Vaquerizas, J., et al, A
census of human transcription factors: function, expression and evolution,
Nature Reviews Genetics 10, 252-263 (April 2009) | doi:10.1038/nrg2538.
16.
Watson, J., et al,
Molecular Biology of the Gene, 5th Ed, Benjamin (San Francisco)
2004.
[1]
See Marks, Chapter 13.
[2]
See Broad https://www.broadinstitute.org/education/glossary/transcription-factor
and Watson et al 544-555. Also http://www.nature.com/scitable/definition/general-transcription-factor-transcription-factor-167 Also from Vaquerizas:
“Transcription factors are key cellular components
that control gene expression: their activities determine how cells function and
respond to the environment. Currently, there is great interest in research into
human transcriptional regulation. However, surprisingly little is known about
these regulators themselves. For example, how many transcription factors does
the human genome contain? How are they expressed in different tissues? Are they
evolutionarily conserved? Here, we present an analysis of 1,391 manually
curated sequence-specific DNA-binding transcription factors, their functions,
genomic organization and evolutionary conservation. Much remains to be
explored, but this study provides a solid foundation for future investigations
to elucidate regulatory mechanisms underlying diverse mammalian biological
processes.”
[3]
From NCBI we have: The protein encoded by this gene, cMYC, is a
multifunctional, nuclear phosphoprotein that plays a role in cell cycle
progression, apoptosis and cellular transformation. It functions as a
transcription factor that regulates transcription of specific target genes. Mutations,
overexpression, rearrangement and translocation of this gene have been
associated with a variety of hematopoietic tumors, leukemias and lymphomas,
including Burkitt lymphoma. There is evidence to show that alternative
translation initiations from an upstream, in-frame non-AUG (CUG) and a
downstream AUG start site result in the production of two isoforms with
distinct N-termini. The synthesis of non-AUG initiated protein is suppressed in
Burkitt's lymphomas, suggesting its importance in the normal function of this
gene. See http://www.ncbi.nlm.nih.gov/gene/4609
[4]
See Goss and Kahn, pp60-61.
[8]
See Marks p 318. Note the colors are also descriptive and do not reflect any
specific RNA base pair pairing. Just as with DNA we would expect similar
bonding of CG and A and U.
[9] It
is worth examining the McGarty DRAFTs on Prostate Cancer and Melanoma to see
this in some detail.


