Cancer metastasis has generally considered that cancer is
clonal, namely it starts with a single cell and that cell starts a process that
laves its local environment and through a process of continual change manages
to metastasize throughout the body. We examine a recent paper by Gundem et al
which examines the metastatic behavior of prostate cancer and as a result of
GWAS they show that it can be poly clonal and continually changing.
We the return to a paper we prepared well over a year ago
regarding Cancer Dynamics and show that in that paper we had not only
anticipated this but more. Although that paper does not yet treat epigenetic
factors, nor does Gundem et al, it can be readily modified to do so.
The results have some significant consequences. Mostly is
the treatment of such cancers. Namely if we have polyclonal metastatic
propagation then pathway methods may have to be multifaceted, namely dealing
with the multiplicity of differing pathway anomalies.
Recent Research
In a recent paper by Gundem et al the authors describe an
analysis they have performed on metastatic prostate cancer cells in a group of
patients. Their general conclusions seem to be two fold; (i) that there are
certain metastases that are polyclonal, namely there are multiple cells
initiating the process, (ii) that the progression of the metastases is complex
with ever increasing changes in genetic expression.
Gundem et al state:
By plotting the cancer
cell fractions of mutations from pairs of samples, we determined the clonal
relationship between the constituent subclones and found evidence for
polyclonal seeding of metastases,
This is a powerful observation. Their approach was in simple
terms to do genome wide analysis and doing so over a set of metastatic
locations. Then using a clustering method they could determine with reasonable
accuracy the clonal and polyclonal results as well as the progression.
Specifically:
Using whole-genome
sequencing, we characterized multiple metastases arising from prostate tumours
in ten patients. Integrated analyses of subclonal architecture revealed the
patterns of metastatic spread in unprecedented detail. Metastasis-to-metastasis
spread was found to be common, either through de novo monoclonal seeding of
daughter metastases or, in five cases, through the transfer of multiple tumour
clones between metastatic sites. Lesions affecting tumour suppressor genes usually
occur as single events, whereas mutations in genes involved in androgen
receptor signalling commonly involve multiple, convergent events in different metastases.
Our results elucidate in detail the complex patterns of metastatic spread and
further our understanding of the development of resistance to
androgen-deprivation therapy in prostate cancer ... We identified a set of high-confidence substitutions,
insertions/deletions, genomic rearrangements and copy number changes present in
each tumour sample….
They conclude as follows:
Our analyses allow us
to view with unprecedented clarity the genomic evolution of metastatic prostate
cancer, from initial tumorigenesis through the acquisition of metastatic
potential to the development of castration resistance. A picture emerges of a
diaspora of tumour cells, sharing a common heritage, spreading from one site to
another, while retaining the genetic imprint of their ancestors. After a long
period of development before the most recent complete selective sweep,
metastasis usually occurs in the form of spread between distant sites, rather than
as separate waves of invasion directly from the primary tumour. This
observation supports the ‘seed and soil’ hypothesis in which rare subclones
develop metastatic potential within the primary tumour, rather than the theory
that metastatic potential is a property of the primary tumour as a whole.
Transit of cells from one host site to another is relatively common, either as monoclonal
metastasis-to-metastasis seeding or as polyclonal seeding. Clonal
diversification occurs within the constraining necessity to bypass ADT, driving
distinct subclones towards a convergent path of therapeutic resistance.
However, the resulting resistant subclones are not constrained to a single host
site. Rather, a picture emerges of multiple related tumour clones competing for
dominance across the entirety of the host.
The challenge in the above analysis is to note as we had in
Cancer Dynamics that as the genetic profile of the cancer cells change, there
is a survival of the fittest occurring, namely a certain cell tries to
dominate, and there is also the issue of stem cells and stem cell control and proliferation.
The issue is one of understanding just what constitutes metastatic growth.
Clearly the cells are in a steady state of genetic change, altering in a survival
based manner to dominate.
Figures 3 and 4 of the paper are the most significant. In
Figure 3 we see depicted the evolving changes in gene structure in clonal and
polyclonal mets. In Figure 4 we see the same in a Nuclear Medicine scan showing
the mets. We show that Figure from Gundem et al below since it is of such
significance.

The above shows the mutations or gene expression alterations
and as they progress. This is a complex but quite important description of the
process. (NOTE: The above is Figure 3 as modified from Gundem et al, Nature,
2015).
As Shen states in a Nature commentary on the Gundem et al
paper:
Next-generation DNA
sequencing technologies have made it apparent that primary tumours are not
clonal (consisting of a single population of genetically identical cells). Instead,
they are composed of subclones, subpopulations of genetically identical cells
that can be distinguished from other subclones by the mutations they harbour.
Such subclones compete for dominance during cancer progression, and drug
treatment can lead to formerly minor tumour subclones becoming dominant if they
are resistant to treatment. Thus, clonal evolution shapes the properties of
tumours and can explain their plasticity in response to therapy. Until now, however,
clonal evolution has not been explored in detail in the context of metastasis…..Taken
together, the current studies might explain why, given the prevalence of
circulating tumour cells in patients with solid tumours, successful metastasis
is relatively rare — metastasis may be facilitated by seeding by cell clusters
containing cooperating clones with distinct properties. If so, it is attractive
to speculate that disseminated single cells could remain dormant until
reawakened by interaction with a cooperative metastatic cell arriving at the
same secondary site. Such a model has the potential to revise our conception of
the properties of tumour-initiating cells, as well the metastatic niche, and
may have implications for therapeutic strategies. For example, understanding
the signalling pathways that mediate such clonal cooperativity may lead to
effective therapies using drugs that target these pathways.
The signally pathway issue is a complex one especially since
we know that suppressing one pathway may excite another. The problem will be
targeting all of the cells.
Previous Work
We have considered this before when we wrote a detailed
paper in 2013 on Cancer Dynamics. In our analysis we examined a set of
continually changing cancer cells, and we further assumed that any cell may
have changed to a cancer cell. We then further assumed a diffusion/flow model
for the propagation of those cells and at the same time assumed a continual
process of genetic change. We also assumed that we could find an organ specific
environment which may be most favorable to growth via ligand/receptor combinations.
Finally we also assumed that cell to cell communications could facilitate the
process. We did not consider at that time any epigenetic factors.
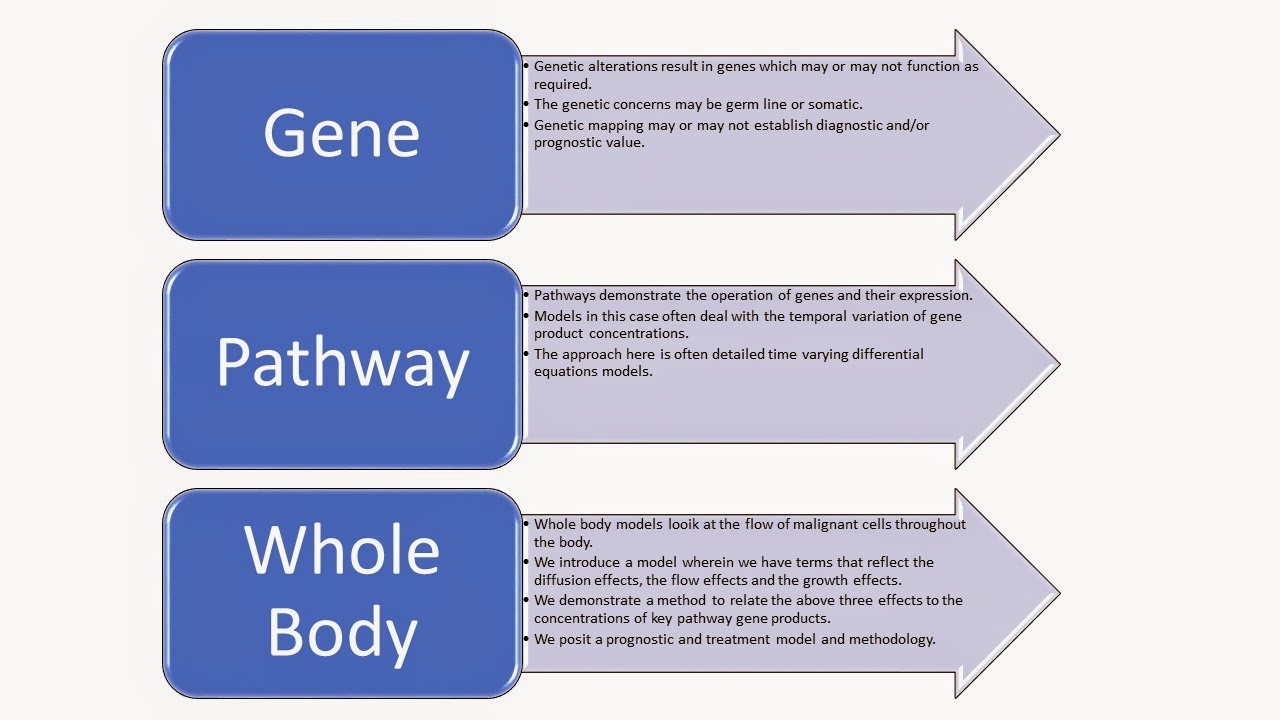
Namely when considering cancer propagation we must consider
the genes, the pathways and the whole body. It is a complex process which we
had developed in the referred to paper.
The equations for the propagation over space and time for a
specific type of cell containing a specific genetic makeup has been shown
below. Here n(x,t) is the concentration or density of a specific cell type, let
us assume a malignant prostate cancer cell, and with a specific genetic
profile. If we examine the Gundem paper we see that this is what they are
looking at from the perspective of a GWAS study of metastatic PCa. However we
have already developed a model and further we had developed an identification
process to provide the drivers in the model itself. Note below that the general
equation is a diffusion plus flow model, diffusion due to evolving
concentrations and flow due to movement within the body itself such a blood
flow dissemination.

The L value are operators and the others are constants
determined in the paper. Our model then allows for polyclonal development and
moreover a complex cell to cell growth stimulus as well
Observations
Now Cancer UK comments on this work as follows[1]:
The team has already
revealed a huge amount of genetic diversity between cancer cells taken from
different sites within each man’s prostate…this new study shows that, despite
the diversity, prostate cancer cells that break free from the tumour and spread
share common genetic faults unique to the individual patient.
Study author … said:
“We gained a much broader view of prostate cancer by studying both the original
cancer and the cells that had spread to other parts of the body in these men.
And we found that all of the cells that had broken free shared a common
ancestor cell in the prostate. The common faults we found in each man could
potentially offer new targets for treatment. But we found that, once cancer
cells have spread, they continue to evolve genetically, so choosing the most
effective treatments will remain a key challenge.”
“The diversity we’ve
found suggests multiple biopsies might be needed to identify the ‘trunk’ of the
cancer’s tree of mutations – we need treatments that target these core
weaknesses to destroy all cancer cells in a clean sweep, rather than trimming
the branches. We must also study more patients to learn how to apply these
findings to develop more personalised treatments for people with the disease.”
“In the phylogenetic
trees that our data have produced, we see that most of the oncogenic mutations
are shared clonally by all the tumour sites in each patient. This common
genetic heritage is a potential achilles heel of the metastases, however, many
of these shared mutations are in tumour suppressor genes and our approach to
therapeutically targeting these needs to be prioritised.
“It takes a while
before a tumour develops the ability to metastasise but once it does the
patient’s prognosis changes significantly. We have to zoom in on this crucial
junction and gather more data on the impact different therapies have on
prostate cancer’s evolution and spread.”
Moreover there are many more concerns. For example:
1. Epigenetic Factors: The analysis does not appear to deal
with the epigenetic factor such as methylation, miRNAs, lncRNAs and the like.
We clearly know that they also have significant impact.
2. Stem Cell Issues: There is also the issue of the stem
cell. Is there such a factor included in or includable in this analysis?
3. Pathway Modifying Therapeutics: As discussed by one of
the commentators the therapeutic implications are evident but in our opinion
not at all clear.
4. Prognosis Analysis: Here we have a significant concern.
Many prognostic tests have been developed. However if we examine for one gene
profile are we missing many others due to poor sampling. Namely one type of
polyclonal cells may be in the profile match but another may not. How, then
does this observation impact the many PCa prognostic profiles out today?
References
1.
Gundem et al, The evolutionary history of lethal
metastatic prostate cancer, Nature 2015. doi:10.1038/nature14347
2.
McGarty, T., Cancer Cell Dynamics, Telmarc, TWP
January 2014, https://www.researchgate.net/publication/271907544_Cancer_Cellular_Dynamics
3.
Shen, M., The complex seeds of metastasis,
Nature, 2015, doi:10.1038/nature14377
[1]
http://www.cancerresearchuk.org/about-us/cancer-news/press-release/2015-04-01-scientists-drill-down-to-genetic-root-of-prostate-tumour-development
also see http://www.sciencedaily.com/releases/2015/04/150401161514.htm
and http://www.sciencedaily.com/releases/2015/04/150402114659.htm
and http://scienceblog.cancerresearchuk.org/2015/04/04/news-digest-prostate-cancer-family-tree-pineapples-walnuts-and-more/


