The main driver for this analysis is the recent work of Chen
and Mellman. In a recent paper these two authors state:
Immunotherapy is proving to be an effective therapeutic
approach in a variety of cancers. But despite the clinical success of
antibodies against the immune regulators CTLA4 and PD-L1/PD-1, only a subset of
people exhibit durable responses, suggesting that a broader view of cancer
immunity is required. Immunity is influenced by a complex set of tumour, host
and environmental factors that govern the strength and timing of the anticancer
response. Clinical studies are beginning to define these factors as immune
profiles that can predict responses to immunotherapy. In the context of the
cancer immunity cycle, such factors combine to represent the inherent
immunological status — or ‘cancer–immune set point’ — of an individual.
The concept of a "set point" is in our opinion
rather poorly used. The construct, if properly understood, means that there is
some point at which T cell activators and inhibitors either permit activation
and effective T cell immunotherapeutic action or inhibit that. Namely there is
some set of activations less inhibitions which all T cells to perform and under
that "set point" they no longer function.
If such a concept has physical meaning, then the authors
state:
Although largely conceptual, the idea of a set point
provides a framework to help organize the torrent of clinical and biomarker
data that will emerge over the coming months and years. The number of targets
that could prove effective for cancer immunotherapy is great; the number of
potential combinations of therapeutic agents that are directed against these
targets (or combinations of such agents with conventional standard-of-care agents)
is even greater. The development of some cancer therapies may be largely
empirical, but it can be guided by considering, even in general terms, the
elements that comprise cancer immunity
Thus, our objective in this paper is to examine this set
point concept and explore its dimensions. Specifically, we examine how it may
be used for therapeutic uses.
To best understand some of the principles we examine some
simplistic model.
Let us begin with a simplistic but representative set of
examples. We know that a CD8 T cell has a receptor, the TCR, T cell receptor,
and it examines an antigen presenting cell, APV, which presents an antigen on
its MHC I protein. This process is essentially an activator process. If that were
all which was needed, then the T cell would be activated and sent out its
cytokines and destroy the cell. However, there are also inhibitor ligands which
activate inhibition in the T cell. Now if the T cell is activated and the inhibition is not
then we get the T cell sending out cytokines and killing the offending cell.
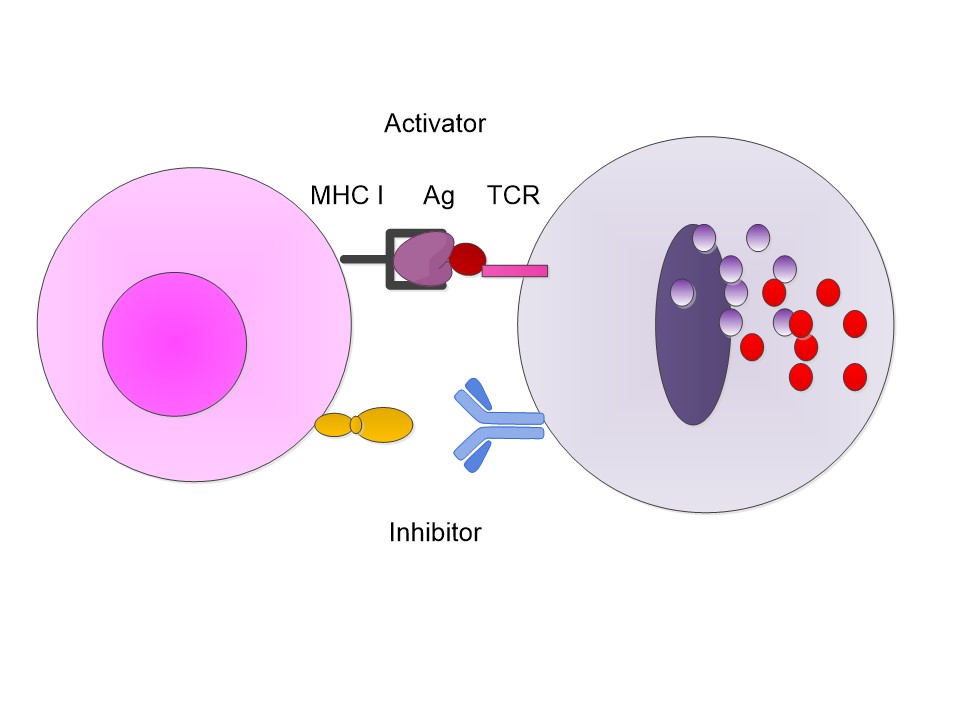
However, if the T cell has an inhibitor also connected then
the inhibitor sends out an internal T cell signal which stops the release. The
presenting cell survives.

Now in reality the T cells do not have just one receptor. It
may have a multiplicity over the surface. Thus, there are a multiplicity of
activators and inhibitors. It is a multiplicity amongst the same type as well a
multiplicity of types. In fact, the T cell may be just covered with receptors
searching out antigens.

Thus, the first layer of complexity of the immune response is
not a simple activator/inhibitor complex but a mass of receptors and ligands
interacting in a complex manner. The question then is; at what point does the T
cell go from active to inactive and back again? In fact, we may ask if there is
some hysteresis effect. If so can we therapeutically take advantage of it.
Ligands and receptors are basically two separate chemical
elements. The binding of them is also essentially a chemical process whereby
the ligand finds the correct location on the receptor to bind. It is in many
ways like any chemical reaction. As such there is a reaction rate whereby the
ligand and receptor combine, but equally there is the reverse reaction, the
breaking apart of a bond.
In the work by Stone et al the authors note:
The interaction between the T-cell receptor (TCR) and its
peptide–major histocompatibility complex (pepMHC) ligand plays a critical role
in determining the activity and specificity of the T cell. The binding
properties associated with these interactions have now been studied in many
systems, providing a framework for a mechanistic understanding of the initial
events that govern T-cell function. There have been various other reviews that
have described the structural and biochemical features of TCR: pepMHC
interactions. Here we provide an
overview of four areas that directly impact our understanding of T-cell
function, as viewed from the perspective of the TCR: pepMHC interaction: (1) relationships between T-cell activity and TCR: pepMHC
binding parameters, (2) TCR affinity, avidity and clustering, (3) influence of coreceptors on pepMHC binding by TCRs
and T-cell activity, and (4) impact of TCR binding affinity on antigenic peptide
specificity.
Now they conclude:
The binding properties of TCRs for their pepMHC ligands
are critically important in the function of T cells, leading to outcomes that
can involve T-cell selection in the thymus or full peripheral T-cell responsiveness
or homeostatic T-cell proliferation in the periphery. The processes are even
more complicated because the same TCR could interact with multiple pepMHC
ligands on the same antigen-presenting cell, each with heterogeneous binding
properties. These reactions would result in a complex integration of signals
that ultimately determine the nature of the T-cell response. While there have been numerous studies to elucidate the
precise binding parameters that correlate with different T-cell activities,
various questions remain unanswered (in part because of the technical
difficulties associated with performing binding experiments on low-affinity
reactions). Further understanding of the TCR binding properties that generate
defined signals is important, not only from a basic science perspective but
also toward developing optimal strategies that improve T-cell responses to foreign
antigens and tumour antigens.
Thus, one must be careful in developing an immune set point
theory to be cautious about the affinity issues as discussed above.
We have examined the complex process fundamentally as a
build. Specifically:
1. Activation: When an antibody binds with the TCR we expect
a response.
2. Inhibitor: When there is an inhibitor, however, it may be
possible to block the pathway leading to the activation.
3. Notwithstanding the above, the cell actually has a multiplicity
of the previous two and thus there may be some race with a finish line defined
by what has been called a "set point", or simply some collection of
activators and inhibitors seeing which one dominates.
4. There are not just one possible activator and inhibitor.
For a T cell, we have the TCR but we may have well more than a PD-1. New
inhibitors are arising each day.
5. The internal machinations of the cellular pathways may
also effect the net result. Thus, genetic changes can affect what happens.
6. The kinetics of the binding can and often do play a
significant role. Binding is not a one-way street, and the result may be loss
of tumor control.
7. Exogeneous Factors: The human biome is often a driving
factor to the efficacy of immunotherapy.
As Chen and Mellman note:
Factors that are extrinsic to the tumour or host genomes
may also affect the immune profile of tumors. Chief among these is the gut
microbiome, which has an important role not only in influencing the initiation
of some cancers, but also in the response to chemotherapy and immunotherapy…mice
bearing subcutaneous syngeneic tumors do not respond to chemotherapy if
sterilized by prior treatment with antibiotics or when raised in germ-free
conditions. The effect was attributed to the ability of commensal bacteria to
activate the innate immune system of the host following chemotherapy, possibly
by causing symbiosis and penetration of commensal bacteria into the gut lamina
propria. Subsequent work established an even clearer link between
T-cell responses and an intact microbiota1. Fecal transfer or co-housing
experiments in mice demonstrated that defined species of gut bacteria enabled
antitumor responses after treatment with anti-PD-L1/PD-1 or anti-CTLA4
therapies. Furthermore, the gut microbiota even influenced spontaneous
antitumor responses, which correlated with the degree of T-cell infiltration
into tumors before any therapy had been administered.
Each of these elements can be considered as a therapeutic
target for immunotherapy. We summarize some of these below.


Thus, understanding the specifics may be a useful approach
in guiding therapeutic development using the immune system.
The previous brief summary lays out some of the issues
inherent in the Chen and Mellman paper. To better understand let us now return
to the ideas of Chen and Mellman. Specifically, their definition of a "set
point". They state:
The cancer–immune set point is the threshold that must be
overcome to generate effective cancer immunity. The set point can be understood
as a balance between the stimulatory factors (Fstim) minus the inhibitory
factors (Finhib), which together must be equal to or greater than 1, over the
summation of all T-cell antigen receptor (TCR) signals for tumour antigens. The
cancer–immune set point is shown here:
∫ (Fstim) − ∫ (Finhib) ≥ 1 ∕ Σ n=1, y (TCRaffinity ×
frequency)
The set point is defined by the summation of the
frequency of peptide–MHC–TCR interactions and TCR signalling in all anticancer
CD8+ T-cell clones (mainly, the TCR affinity for the antigen–MHC class I
complex) against antigens present in the cancer cells, including neoantigens
and cancer-associated antigens, and the endogenous balance of the positive and
negative immune regulators that are inherent to each host or patient.
Now just what this means is somewhat open for debate because
it is written by a biologist not a physical scientist and definitely not an
engineer. Permit me to attempt an interpretation. First let us try to be
specific about a definition. Namely some definition of a variable which is
measurable.
Let us try to first understand the F terms.
Fstim: This is a stimulatory factor. What is it? One could
guess it is some cell with an MHC I presenting some antigen Ag to a T cell
receptor TCR. Should we examine cell by cell? Should we look at every possible
T cell, namely ones that say are CD 8 T cells, or how about other immune cells.
Why not include NK cells as well? Should we look at stem cells only, do we know
what they are? Do we then count these for every T cell, for a mass of T cells,
for what?
Finhib: We know some of these we believe. There is PD-1 and
CTLA-4. They can block the T cell from attacking. We also suspect that there
are many others we have yet to find. So, let us simplistically assume we can
model with the two mentioned. But what are we measuring? Are we measuring a
single cell, a collection of cells, the totality of all cells? Are we measuring
all stimulatory factors or just a few? Are we measuring all inhibitory factors
or just the ones we know? Are we weighting some differently than others or the
same?
This if we have two single cells and it has say 50 T cell
receptors and 45 PD-1 receptors, then we can have activated say 35 of the TCR
and have activate say 22 of the PD-1. Now what happens? Is activation by each
TCR the same and can a TCR being activated be inhibited by an activated PD-1 on
a one to one basis?

The above still has no physical meaning. Now let us consider
T Cell Affinity. As Nicholich et al state:
Affinity refers to the steady-state association constant
between a monovalent receptor and its ligand, in this case a single T-cell
receptor (TCR) and peptide–MHC (pMHC) complex. Structural avidity is the
steady-state association constant between multiple cell-bound receptors and
ligands and is determined by the direct binding affinities of multiple TCRs to
their pMHC complexes. Functional avidity depends on the relative kinetics of
signalling that translate into measurable biological functions such as
proliferation, cytokine production or cytolytic function. APC,
antigen-presenting cell.
Now as Hsieh et al note:
TCR affinity: The strength of interaction between the T
cell receptor and a single peptide–MHC complex.
As an abstraction that may be fine but as something used in
a measurement and equation it is highly deficient.
Now as Daniels et al note:
To estimate the TCR affinity of the ligands comprising
the selection boundary, we measured tetramer binding; which correlates with
monomeric TCR–pMHC affinities, is performed on live cells and involves the
participation of CD8. The binding characteristics of tetramers were determined
on pre-selection OT-I double positive thymocytes at 37 uC. The dissociation
constant (Kd) was calculated by nonlinear regression analysis and confirmed by
homologous competition experiments. The tetramer binding curves for Q4R7 (weakest negative
selector), T4 (border ligand) and Q4H7 (strongest positive selector)
overlapped. Their Kd values (Q4R7, 4869.5 nM; T4, 55610.1 nM; Q4H7, 5169.1 nM;
n57, P50.455) and their half-lives (t1/2) were not significantly different
(Table 1). However, heterologous competition assays showed that Q4R7 was more
efficient than Q4H7 at inhibiting the binding of OVA tetramers.
or perhaps they mean something akin to this:

Now we know that there is a threshold effect for activating
and suppressing. Namely there has to be more activators than suppressors. Just
what that balance is of course is uncertain. Again, the statement has no
physical meaning.
They continue:
This can be further influenced by other elements of
immunity, including tumour-derived immunomodulatory components, as well as by
exogenous factors such as infection and exposure to pharmacological agents. A
given patient with cancer may have a low set point, making it easier to
generate an anticancer immune response, or a high set point, which makes it
more difficult.
The aim of immunotherapy is to increase Fstim, decrease
Finhib or increase TCR signalling to drive progression of the cancer-immunity
cycle. These values are difficult to quantify with current techniques but
represent a useful theoretical construct. It is probable that the cancer–immune
set point of a particular person is already determined by the time of clinical
presentation, driven by the inherent immunogenicity of the tumour and by the
responsiveness of the individual’s immune system. Although it is reasonable to assume that various lines of
cancer therapy or changes in environmental factors might alter Fstim and
Finhib, such changes might only be transient. Often, the set point that is
identified using pretreatment biopsies is similar to the set point determined
by biomarker profiling from biopsies taken on progression after therapy. Likewise, despite the continued accumulation of mutations
in a tumour as a function of time, primary and metastatic lesions can exhibit
similar immune profiles. The features that determine the set point may
therefore reflect genetic factors that are specific to a given tumour, the
genetics of the person with cancer, or the extent to which antitumor immunity
had developed initially. Conceivably, immunotherapy may work as a consequence
of either its direct effect on Fstim and Finhib (that is, by assisting the
completion of a single revolution of the cancer-immunity cycle) or its ability
to alter the set point (for example, by propagating the cancer-immunity cycle,
which enhances the cancer-specific T-cell response). Although largely conceptual, the idea of a set point
provides a framework to help organize the torrent of clinical and biomarker
data that will emerge over the coming months and years. The number of targets
that could prove effective for cancer immunotherapy is great; the number of
potential combinations of therapeutic agents that are directed against these
targets (or combinations of such agents with conventional standard-of-care
agents) is even greater.
Thus, let us
try and construct meaning which may be measurable and verifiable as well as
actionable. Consider the following model:
1. Let us assume we have a tumor cell. Let us assume there
are N possible activator ligands and M inhibitor ligands.
2. Let us assume that for each of the above ligands we have
on a T cell some receptor. If there is a ligand without a receptor we shall
ignore it.
3. Assume we can count and differentiate the differing
ligand-receptor possibilities on a cell.
4. Now calculate the following:

Namely, we count the number of different activators and the
number of different inhibitors and then weight them by some metric, yet to be
determined, and then weigh them by the total present.
This approach may have merit. The weights may be unity, but
that is a mere guess. The weights may be reflective of the enzymatic
consistency of the contact. Frankly we just do not know but it is worth
exploring.
We now return to following Chen and Mellman and their
observations. They note:
The role of the immune system in cancer remained
unappreciated for many decades because tumors effectively suppress immune
responses by activating negative regulatory pathways (also called checkpoints)
that are associated with immune homeostasis or by adopting features that enable
them to actively escape detection. Two such checkpoints, cytotoxic T-lymphocyte protein 4
(CTLA4) and programmed cell death protein 1 (PD-1), have garnered the most attention
so far. CTLA4 is a negative regulator of T cells that acts to
control T-cell activation by competing with the co-stimulatory molecule CD28
for binding to shared ligands CD80 (also known as B7.1) and CD86 (also known as
B7.2). The cell-surface receptor PD-1 is expressed by T cells on
activation during priming or expansion and binds to one of two ligands, PD-L1
and PD-L2. Many types of cells can express PD-L1, including tumour cells and
immune cells after exposure to cytokines such as interferon (IFN)-γ; however,
PD-L2 is expressed mainly on dendritic cells in normal tissues. Binding of
PD-L1 or PD-L2 to PD-1 generates an inhibitory signal that attenuates the
activity of T cells. The ‘exhaustion’ of effector T cells was identified
through studies of chronic viral infection in mice in which the PD-L1/PD-1 axis
was found to be an important negative feedback loop that ensures immune
homeostasis; it is also an important axis for restricting tumour immunity.
They then proceed to characterize three differing states of
tumors with respect to their T cell response. They are:
1. Inflamed Tumor: This a tumor with lots of cells and
penetrating the tumor space.
2. Immune Desert Tumor: This is a tumor with lots of cells
but no significant penetration of the tumor space.
3. Immune Excluded Tumor: This is a tumor with a paucity of
any T cells present.
Now we consider the descriptions as presented by Chen and
Mellman:
The first profile, the immune-inflamed phenotype, is
characterized by the presence in the tumour parenchyma of both CD4- and
CD8-expressing T cells, often accompanied by myeloid cells and monocytic cells;
the immune cells are positioned in proximity to the tumour cells. Samples from
inflamed tumors may exhibit staining for PD-L1 on infiltrating immune cells
and, in some cases, tumour cells. Many proinflammatory and effector cytokines
can also be detected by mRNA analysis in these sections of tumors. This profile
suggests the presence of a pre-existing antitumor immune response that was
arrested probably by immunosuppression in the tumour bed. Indeed, clinical
responses to anti-PD-L1/PD-1 therapy occur most often in patients with inflamed
tumors…The second profile is the immune-excluded phenotype,
which is also characterized by the presence of abundant immune cells. However,
the immune cells do not penetrate the parenchyma of these tumors but instead
are retained in the stroma that surrounds nests of tumour cells. The stroma may
be limited to the tumour capsule or might penetrate the tumour itself, making
it seem that the immune cells are actually inside the tumour. After treatment
with anti-PD-L1/PD-1 agents, stroma-associated T cells can show evidence of
activation and proliferation but not infiltration, and clinical responses are
uncommon. These features suggest that a pre-existing antitumor response might
have been present but was rendered ineffective by a block in tumour penetration
through the stroma or by the retention of immune cells in the stroma. T-cell
migration through the tumour stroma is therefore the rate-limiting step in the
cancer–immunity cycle for this phenotype.
Finally, the third type is characterized as follows:
The third profile, the immune-desert phenotype, is
characterized by a paucity of T cells in either the parenchyma or the stroma of
the tumour. Although myeloid cells may be present, the general feature of this
profile is the presence of a non-inflamed tumour microenvironment with few or
no CD8-carrying T cells. Unsurprisingly, such tumors rarely respond to
anti-PD-L1/PD-1 therapy. This phenotype probably reflects the absence of
pre-existing antitumor immunity, which suggests that the generation of
tumour-specific T cells is the rate-limiting step. The immune-desert phenotype
and the immune-excluded phenotype can both be considered as non-inflamed
tumors.
Thus, this does pose the question; how does one identify
these cells and how could one move one category to the other for better
response? Frankly one asks just what is happening from one class to another?
Set Points, Check Points, and other elements of the control
of the immune system as a mechanism to understand and deal with cancer has been
evolving at a rapid pace. Where the Check Point field seeks new and effective
ligand-receptor pairs, the Set Point field seems to examine the process in a
more holistic manner. Perhaps that is an approach which would enable a more
systematic approach.
How does this process change as a cell matures? What of cell
differentiation. T cells like many of the lymphoid line go through varying
degrees of maturation. Thus, we ask: what is the difference?
We have discussed the stem cell constructs at length. In
McGarty (Stem Cells) we have tried to bring some of these ideas up to data. The
problem is that stem cells may very well have different markers than the cells
we can attack with the tools at hand. Thus, attacking PD-1 and CTLA4 markers
may work for the mass of the tumor and result in shrinkage but it may totally
miss the stem cell. How best to address this is uncertain?
What are the therapeutic dimensions of this principle? We
have discussed a few here but there are many which present themselves.
CAR-T cells are "engineered" T cells which are
designed by use of such tools as a lentivirus to attack a specific malignant
cell. They have been shown to be useful for hematological cancers and have been
examined for solid tumors. As Ramachandran notes in his Thesis:
As the name suggests, a CAR is a chimera of domains from
different proteins assembled together to create a functional receptor. These
novel receptors initiate a functional downstream effector T-cell signaling
pathway when they encounter target antigen, usually the TAA on a cancer cell.
This gives the opportunity to engineer a large variety of TAA-specific
receptors targeting a broad range of cancer types.
CARs typically contain four domains
(a) extracellular antigen binding domain: It confers the
antigen-specificity to the engineered T-cell. A majority of the engineered CARs
for cancer therapy have antibody-derived antigen binding domains called
single-chain variable fragment (scFv). CARs containing a scFv extracellular
domain retain the specificity of an antibody. A major advantage of having scFv
extracellular domain is that it bypasses the need for antigen presentation by MHC-I
on tumor cells, as antibodies directly bind to cell surface antigens. (b) Spacer or hinge region: It gives flexibility and
length to allow proper dimerization of scFv, thus improving its stability. The
most commonly used spacer regions are derived from IgG Fc CH2-CH3 domains, CD28
hinge domain and CD8α spacer domain (c) Transmembrane domain: It determines the stability of
CAR expression on cell surface. The most commonly used transmembrane regions
are derived from CD3ζ CD4, CD8 and CD28 molecules138 and (d) Cytoplasmic signaling domain(s): This region has the
domains that provide the necessary downstream signaling for T-cell effector
functions. CARs are classified into different generations based on
the number of cytoplasmic signaling domains namely first, second and third
generation CARs. First generation CARs have only one cytoplasmic domain,
usually T-cell activation signaling domain (CD3ζ chain). In addition to the
T-cell activation domain second generation CARs have one extra co-stimulatory signaling
domain, e.g., CD28, 4-1BB, ICOS or OX40 and third generation CARs have two
extra co-stimulatory domains…
In a recent Technical Note McGarty has further developed the
CAR-T cell concepts for both hematological and solid tumors. CAR-T are engineered
to specific targets. The question then is; can a better understanding of set
points allow for improved targeting for CAR-T cells or are CAR-T cells perforce
of their design not really useful for attacking solid tumors?
The enzyme kinetics of the reactions on the surfaces of T
cells and APC or tumor cells are critical. We have almost always assumed that
once a protein is bound it stays. Yet we know it is not the case. Furthermore,
when understanding the set point model, if we have a paucity of activators on a
T cell it will not function. If the paucity is due to enzymatic action, then
perhaps we can indirectly address the low level by increasing the retention via
enzyme kinetic improvement.
The pathway factors are both integral to immunotherapeutic
approaches, they facilitate the process inside a T cell for example, but they
may also be poorly understood. Let us briefly review that issue. We must look
at pathways from the perspective of the T cell and the tumor Ag presenting
cell.
1. From the T cell perspective we have internal genetic
pathways which facilitate the process of cytokine release. If there are faults
on the pathway, then we would not expect the T cell to function. Thus, we may
ask if these are somatic defects or a result of some change in the T cell.
2. From the perspective of the tumor cell, we know its
pathways have usually been altered. Then does this altering result in the
excess expression of inhibitors or the suppression of activators. Do the pathways
alter the MHC I presentation efforts?
Both dimensions are worth examining.
A recent National Academies Report by Balogh et al present
several policy issues regarding immunotherapy. The report was meant to present
a simplified overview of immunotherapeutics as well as present some key policy
issues. Concerns regarding costs, patient value, physician-patient expectations
were discussed. Also, was a discussion
on evidence based approaches. The problem is that the experience is limited and
the costs high. Furthermore, what seemed not discussed was the fact that the
complexity of this field is great and the depth of understanding by physicians
quite limited. One could say that most Oncologists are trained to administer
chemotherapy, and have a limited if not aged understanding of the immune
system.
1.
Balogh, E., et al, Policy
Issues in the Clinical Development and Use of Immunotherapy for Cancer
Treatment: Proceedings of a Workshop, http://www.nap.edu/23497
National Academies Press, 2016.
2.
Caligiuri, M., Human
natural killer cells, Blood, 1 August 2008, Volume 112, Number 3
3.
Chen, D., I. Mellman,
Elements of cancer immunity and the cancer–immune set point, 19 January 2017, Vol
541, Nature, 321
4.
Cornish-Bowden, A.,
Fundamentals of Enzyme Kinetics, Wiley (Weinheim GE) 2012.
5.
Daniels, et al, Thymic
selection threshold defined by compartmentalization of Ras/MAPK signalling,
Nature, Dec 2006.
6.
Dolan, D., S. Gupta, PD-1
Pathway Inhibitors: Changing the Landscape of Cancer Immunotherapy, Cancer
Control, July 2014.
7.
Freeman, G., Structures of
PD-1 with its ligands: Sideways and dancing cheek to cheek, PNAS, July 29, 2008,
vol. 105, no. 30, 10275–10276
8.
Galluzzi, L. et al, Classification
of current anticancer immunotherapies, Oncotarget, Vol. 5, No. 24, 2016.
9.
Hsieh et al, Selection of
regulatory T cells in the thymus, Nature Reviews, Immunology Volume 12, March 2012,
157
10. Ibarrondo, F., et al, Natural Killer T Cells in Advanced
Melanoma Patients Treated with Tremelimumab, PLOS ONE, www.plosone.org, October
2013, Volume 8, Issue 10, e76829
11. Kindt et al, Kuby Immunology, Freeman (New York) 2007
12. Krogsgaard, et al., T cell receptor affinity and avidity defines
antitumor response and autoimmunity in T cell immunotherapy, Journal for
ImmunoTherapy of Cancer 2013
13. McGarty, T. CAR-T Cells and Cancer, https://www.researchgate.net/publication/309419224_CAR_T_Cells_and_Cancer
, 2016.
14. McGarty, T., Cancer Stem Cells and Cancer of Origin Redux, https://www.researchgate.net/publication/301542243_Cancer_Stem_Cells_and_Cancer_of_Origin_Redux
2016.
15. McNeel, D., TCR diversity – a universal cancer immunotherapy
biomarker? Journal for ImmunoTherapy of Cancer (2016) 4:69
16. Nikolich, J., et al, The many important facets of T-cell
repertoire diversity, Nature Reviews Immunology 4, 123-132 (February 2004)
17. Pittari, G., et al, Revving up natural killer cells and
cytokine-induced killer cells against hematological malignancies, Frontiers in
Immunology, www.frontiersin.org, May 2015, Volume 6, Article 230
18. Prendergast, G., E. Jaffee, Cancer Immunotherapy, Academic (New
York) 2013.
19. Ramachandran, M., Cancer Immunotherapy Evolving Oncolytic
viruses and CAR T-cells, PhD Dissertation Uppsala Univ 2016.
20. Sauro, H., Enzyme Kinetics in Systems Biology, Ambrisius
(Seattle) 2013.
21. Schweizer, M., C. Drake, Immunotherapy for Prostate Cancer –
Recent Developments and Future Challenges, Cancer Metastasis Rev. 2014 September;
33(0): 641–655
22. Steer et al, Harnessing the immune response to treat cancer,
Oncogene (2010) 29, 6301–6313
23. Stetson, D., et al, Constitutive Cytokine mRNAs Mark Natural
Killer (NK) and NK T Cells Poised for Rapid Effector Function, J. Exp.
Med. The Rockefeller University Press,
Volume 198, Number 7, October 6, 2003 1069–1076
24. Stone, et al, T-cell receptor binding affinities and kinetics:
impact on T-cell activity and specificity, Immunology, 126, 165–176, 2009.
25. Topalian, S., et al, Immune Checkpoint Blockade: A Common
Denominator Approach to Cancer Therapy, Cancer Cell 27, April 13, 2015
26. Vivier, E. et al, The Example of Natural Killer Cells, 7 January
2011 Vol 331 Science
27. Wojciech, et al, the same self-peptide selects conventional and
regulatory CD4þ T cells with identical antigen receptors, Nature Comm, Oct 1,
2014.
28. Zhang, et al, Direct Measurement of T Cell Receptor Affinity and
Sequence from Naïve Anti-Viral T Cells, Sci Transl Med. 2016 June 01; 8(341)


