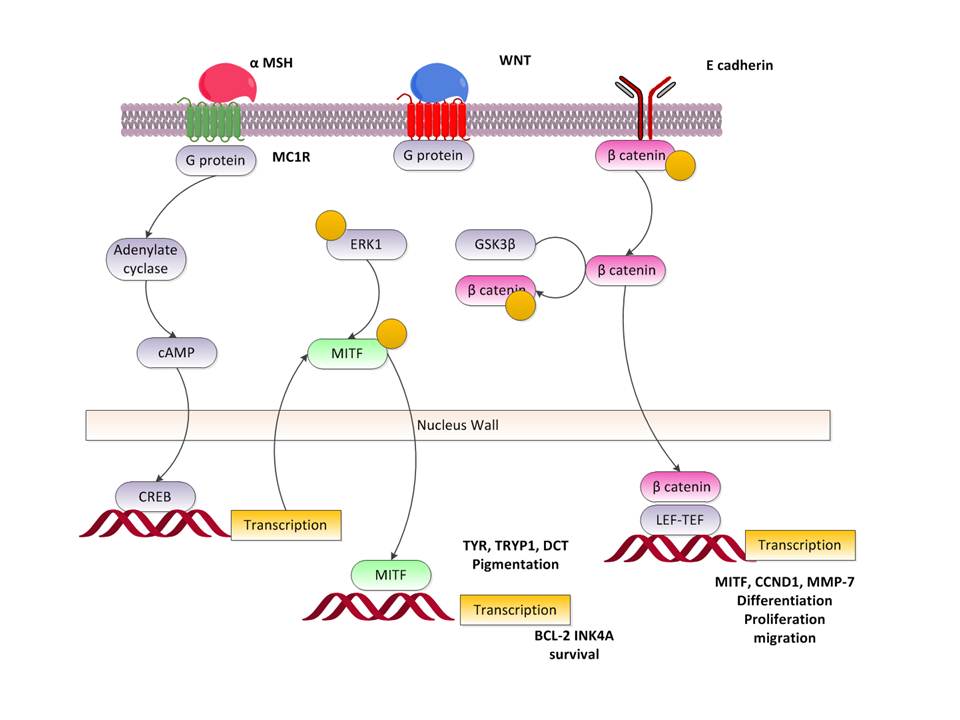
The focus on intracellular pathways has been a prime
direction of research in the development of cancers. However there has from
time to time been some focus on the extracellular matrix, the “ECM”, which
relates in many ways to the stability of the cell, its localization. Cancer
cells lose this sense of localization and begin to move. We have written a
White Paper which details much of this insight and we summarize it here.
The processes at play in the ECM have a significant impact
on the processes that occur within a cell. Thus it is essential to have an
understanding of the ECM. Recent work by Fisher and his people on MDA-9, a
controller of certain ECM elements, demonstrates a control path that influences
the internal pathways. We discuss the ECM in the context of the MDA-9
developments.
In this section we use a recent development in understanding
the impact of Mda-9 and the nexus with the extra cellular matrix, ECM, and the
control of metastatic melanoma.
We first review the Fisher Team efforts as recently
presented and then we examine the standard intracellular pathways that have
been examined and from that we provide an overview of the extra cellular
matrix, ECM, which is the “glue” binding together cells and facilitating cell
to cell communications.
We find this an interesting focus or research for several
reasons:
1. It examines the ECM which has received limited focus.
2. It focuses on pathways as we have been also doing and
specifically an interesting adjunct to the current B-RAF approach.
3. It establishes a clear path forward which is logically
and experimentally based and verifiable.
There has been limited prior research on these issues. In
Hearing and Leong, 380-386, there is a limited discussion regarding the ECM and
melanoma with references. The work by Zent and Pozzi provides a broad and
detailed perspective of the ECM with many cancers. However their work is not
specific to melanoma. In Weinberg there are references but there does not
appear to be any singular focus on the ECM as a standalone system element.
In the recent paper by Das et al, the authors (from Fisher’s
Lab at Virginia Commonwealth) state
:
Melanoma differentiation associated gene-9 (MDA-9), also
known as syntenin, functions as a positive regulator of melanoma progression
and metastasis. In contrast, the Raf kinase inhibitor RKIP, a negative modulator
of RAF-stimulated MEKK activation, is strongly downregulated in metastatic
melanoma cells. In this study, we explored an hypothesized inverse relationship
between MDA-9 and RKIP in melanoma. Tumor array and cell line analyses
confirmed an inverse relationship between expression of MDA-9 and RKIP during
melanoma progression.
We found that MDA-9 transcriptionally downregulated RKIP
in support of a suggested crosstalk between these two proteins. Further, MDA-9
and RKIP physically interacted in a manner that correlated with a suppression
of FAK and c-Src phosphorylation, crucial steps necessary for MDA-9 to promote
FAK/c-Src complex formation and initiate signaling cascades that drive the
MDA-9-mediated metastatic phenotype.
Lastly, ectopic RKIP expression in melanoma cells
overrode MDA-9-mediated signaling, inhibiting cell invasion,
anchorage-independent growth and in vivo dissemination of tumor cells. Taken
together, these findings establish RKIP as an inhibitor of MDA-9-dependent
melanoma metastasis, with potential implications for targeting this process
therapeutically.
From the paper by Houben et al we have the RKIP activation
as shown below:
As Houben et al state:
The Ras/Raf/MEK/ERK intracellular signalling cascade is a
major determinant in the control of cell growth, differentiation, and survival
and can be activated in response to a variety of extracellular stimuli.
Stimulation of growth factor receptors results in the activation of the small
G-protein Ras, which in turn interacts with the protein kinase Raf leading to
its activation. MAP kinase kinase kinase (Raf) phosphorylates and activates MAP
kinase kinase (MEK), and MEK phosphorylates and activates extracellular
signal-regulated kinase (ERK) 1/2 (p42/p44 MAP kinases).
Although Raf and MEK appear largely restricted to only
one class of substrates, ERK targets more than 70 substrates including
membrane, cytoskeletal, cytoplasmic, nuclear, and even mitochondrial proteins.
Recently, a negative regulator of this pathway has been described. The Raf
Kinase Inhibitor Protein (RKIP) binds to either Raf or MEK and thereby
interferes with the activation of MEK by Raf. The importance of the
Ras/Raf/MEK/ERK signalling pathway for carcinogenesis is well established. Indeed,
Ras genes (K-ras, H-ras, and N-ras) are the most frequently
mutated oncogenes detected in human cancer.
Houben et al further state about RKIP (12q24.23) as a target
the following:
To assess the relevance of the Ras/Raf/MEK/MAP kinase
pathway, we analyzed for activating B-Raf mutations and we elucidated the
presence of the Raf Kinase Inhibitor Protein (RKIP) and extracellular
signal-regulated kinase (ERK) as well as the phosphorylation status of ERK. All
MCC samples were negative for the B-RafV600E
mutation. Remarkably, RKIP, which was shown to interfere with the
activation of MEK by Raf, was highly expressed in primary as well as in
metastatic MCC. … Western blot analysis of three MCC-derived cell lines
revealed in one case the pattern present in situ (i.e. high RKIP expression and
complete absence of phosphorylated ERK).
Thus the Fisher team seems to seek out a RKIP inhibitor to
slow the pathway. This is in addition to the B-RAF inhibitors which are
currently in clinical use.
Now in an industry piece on the same article the author Ho
states
:
…. the scientist believes that they have the ability to
eliminate melanoma differentiation associated gene-9 (mda-9)/syntenin, a
specific protein. In the experiment, the researchers discovered that Raf kinase
inhibitor protein (RKIP) was able to interact and suppress with mda-9/syntenin.
The protein was originally cloned in a laboratory and past studies showed how
it interacted with c-Src, another protein, to produce a set of chemical
reactions that later boosted metastasis.
“Prior research suggests that RKIP plays a seminal role in
inhibiting cancer metastasis, but, until now, the mechanisms underlying this
activity were not clear,” explained Paul Fisher, the program co-leader of
Cancer Molecular Genetics at Virginia Commonwealth University Massey Cancer
Center, in a prepared statement. “In addition to providing a new target for
future therapies, there is potential for using these two genes as biomarkers
for monitoring melanoma development and progression.”
The team of investigators discovered that RKIP become
attached to mda-9/syntenin, which resulted in limiting the expression of
mda-9/syntenin. With the finding of this physical interaction, the scientists
believe that they could possibly create small molecules that are similar to
RKIP and the molecules could be used as drugs to treated metastasis in cancers
like melanoma.
We depict this pathway below:
The article continues:
There was also a difference in terms of the level of
mda-9/syntenin and RKIP. While malignant and metastasis melanoma cells had
higher levels of mda-9/sytnenin compared to RKIP, the healthy melanocyte cells
that create pigment in eyes, hair, and skin had higher levels of RKIP than
mda-9/syntenin. The researchers believe that different levels in the proteins
could be used in diagnosis, particularly in following the progression of a
disease or tracking a patient’s response to a particular treatment.
“Our findings represent a major breakthrough in
understanding the genetic mechanisms that lead to metastasis in melanoma. Prior
studies have shown that levels of mda-9/syntenin are elevated in a majority of
cancers, including melanoma, suggesting that our findings could be applicable
for a wide range of diseases,” continued Fisher, who also serves as chairman of
VCU’s Department of Human and Molecular Genetics and director of the VCU
Institutes of Molecular Medicine, in the statement.
Moving forward, the scientists plan to determine how they
can develop small molecules that mimic RKIP. These molecules could potentially
be utilized in new treatments for melanoma.
This is a fundamental result. It demonstrates another
pathway element and at the same time connects the intracellular pathways with
the extra cellular matrix and their pathways. Potentially this is diagnostic,
prognostic and a treatment as well.
The following Figure is a repetition of the standard
intra-cellular pathways. We have discussed these at length.
What is different from what we have detailed previously is
the Extra Cellular Matrix connection via the integrins. This yields the
controlling FAK path using FAK and Src. Note that this activates RTK and Ras
and thus as we have described many of the other internal pathways this is the
first time we have involved the ECM directly. The ECM is a significant element
in cancer proliferation, it is the sea in which the changing cells sail
metaphorically but at the same time it allows communication with the
environment as well as presenting ligands to receptors.
As depicted in Sarkar et al, we have the following sets of
paths and the results:
We shall be examining these in some detail. Let us first
characterize some of the above identified elements controlled by the
extracellular matrix path. The others we have examined in detail elsewhere.
FAK is also known as; PTK2, FADK; FAK1; FRNK; PPP1R71;
p125FAK; pp125FAK. It is located at 8q24.3. It is a kinase.
NCBI states its function as follows:
This gene encodes a cytoplasmic protein tyrosine kinase
which is found concentrated in the focal adhesions that form between cells
growing in the presence of extracellular matrix constituents. The encoded
protein is a member of the FAK subfamily of protein tyrosine kinases but lacks
significant sequence similarity to kinases from other subfamilies. Activation
of this gene may be an important early step in cell growth and intracellular
signal transduction pathways triggered in response to certain neural peptides
or to cell interactions with the extracellular matrix. Several transcript
variants encoding different isoforms have been found for this gene, but the
full-length natures of only three of them have been determined.
SRC is located at 20q12-q13. As noted in NCBI
:
This gene is highly similar to the v-src gene of Rous
sarcoma virus. This proto-oncogene may play a role in the regulation of
embryonic development and cell growth. The protein encoded by this gene is a
tyrosine-protein kinase whose activity can be inhibited by phosphorylation by
c-SRC kinase. Mutations in this gene could be involved in the malignant
progression of colon cancer. Two transcript variants encoding the same protein
have been found for this gene.
The p38 gene has multiple names. It is MAPK14, RK; CSBP;
EXIP; Mxi2; CSBP1; CSBP2; CSPB1; PRKM14; PRKM15; SAPK2A; p38ALPHA. It is
located at 6p21.3-p21.2.
Its function described by NCBI is as follows
:
The protein encoded by this gene is a member of the MAP
kinase family. MAP kinases act as an integration point for multiple biochemical
signals, and are involved in a wide variety of cellular processes such as
proliferation, differentiation, transcription regulation and development.
This kinase is activated by various environmental
stresses and proinflammatory cytokines.
The activation requires its phosphorylation by MAP kinase
kinases (MKKs), or its autophosphorylation triggered by the interaction of MAP3K7IP1/TAB1
protein with this kinase. The substrates of this kinase include transcription
regulator ATF2, MEF2C, and MAX, cell cycle regulator CDC25B, and tumor
suppressor p53, which suggest the roles of this kinase in stress related
transcription and cell cycle regulation, as well as in genotoxic stress
response.
Four alternatively spliced transcript variants of this
gene encoding distinct isoforms have been reported.
We have discussed this before. We reiterate what that
discussion contains. NF-κB is
a transcription factor that resides in the cytoplasm. It is called Nuclear
Factor and was identified by David Baltimore as an enhancer factor for the κ chain of Ig light chain in B
lymphocytes. When activated it moves to the nucleus and is a transcription
factor in activating over 400 genes. It is activated by a large number of
stimuli and its action of a large gene set causes significant DNA activity. NF-κB appears on 10q24 and is somatic
and acts in a dominant manner.
We now depict this putative pathway based upon the work of
Kwang and Aggarwal. This is shown below. Activated NF-κB is clearly an
activator of an anti-apoptosis process in the nucleus. The paper by Huang et al
shows that blockade of NF-κB is an effective suppressor of angiogenesis,
invasion and metastasis of prostate cancer.
Proteins of the matrix metalloproteinase (MMP) family are
involved in the breakdown of extracellular matrix in normal physiological
processes, such as embryonic development, reproduction, and tissue remodeling,
as well as in disease processes, such as arthritis and metastasis. Most MMP's
are secreted as inactive proproteins which are activated when cleaved by
extracellular proteinases.
The enzyme encoded by this gene degrades type IV and V
collagens. Studies in rhesus monkeys suggest that the enzyme is involved in
IL-8-induced mobilization of hematopoietic progenitor cells from bone marrow,
and murine studies suggest a role in tumor-associated tissue remodeling
We shall discuss MMP in detail when we summarize the ECM.
The protein encoded by this gene is a small GTPase of the
Rho-subfamily, which regulates signaling pathways that control diverse cellular
functions including cell morphology, migration, endocytosis and cell cycle
progression. This protein is highly similar to Saccharomyces cerevisiae Cdc 42,
and is able to complement the yeast cdc42-1 mutant.
The product of oncogene Dbl was reported to specifically
catalyze the dissociation of GDP from this protein. This protein could regulate
actin polymerization through its direct binding to Neural Wiskott-Aldrich
syndrome protein (N-WASP), which subsequently activates Arp2/3 complex.
Alternative splicing of this gene results in multiple transcript variants.
The ECM has often been neglected when discussing cancer
pathways. Weinberg has multiple references but does not seem to place it in any
specific spotlight. In Lewin, Cell, the discussion is quite well focused but
yet there is but passing reference to the impact on cancer pathways.
Specifically in Lewin on p 850 there is reference to MMP-9, here a metalloproteinase,
and melanoma
.
The ECM is the collection of molecules that lie between the
cell walls. The ECM provides for structural integrity as well as facilitates
and even participates in cell to cell communications. The ECM is a highly
complex and quite active element in the ongoing life of the cells. In addition
we all too often look to what happens in a cell, with at best a nod to ligands,
and we do not look at the cell internals as well as the ECM as a holistic
system totality. The work of the Fisher Team in a small way may help refocus
this effort on the complex as a working whole.
We will follow Lewin and deal with the principal
participants in the ECM. There are a wealth of books which focus on this area.
Collagens provide structure support. They are triple helical
proteins wrapped to provide that supporting structure between the cells. There
any many types of collagen and the actually assembly commences within the cell
and the semi-finished product passes through the cell wall to the ECM. For our
purposes the collagen complexes are at this time of limited interest.
Fibronectin facilitates the process of connecting cells to
matrices of collagen. Fibronectin proteins have a six element structure. Cells
bind to fibronectin via receptors called integrins. The fibronectin binding
thus activates pathways within the cell, thereby establishing an intra and
intercellular pathway complex. The pathways activated control growth, movement
and cell differentiation.
We can now examine some of the relevant literature on
fibronectin and melanomas. As Yi and Ruoslahti state:
Fibronectin is a prototypic extracellular matrix (ECM)
protein that is deposited by various types of cells into an adhesive fibrillar
meshwork of protein (1). Fibronectin, and ECM in general, control many cellular
functions, including growth, migration, differentiation, and survival. The
signals that control these behaviors are transmitted from the ECM to the cell
by integrins, a family of transmembrane receptors (2, 3). Malignant cells often
bypass the ECM–integrin signaling system; they are not bound by the spatial
constraints imposed by the ECM on normal cells, and they no longer require ECM
contact for survival
Liu et al state:
Tumor cells frequently exhibit decreased adhesiveness due
to failure to deposit stromal fibronectin (FN), permitting more rapid
proliferation, migration, invasion, and metastasis. Although up-regulation of
FN has been noted in gene profiles of carcinomas compared with normal tissue,
reduced FN expression has been described at the peripheral margins of invading
tumors. In this study, we investigate the role of FN in cancer behavior. …
Neoplastic transformation is often characterized by changes in the organization
of the cytoskeleton, decreased cell adhesion, and aberrant adhesion–mediated
signaling (2). Disruption of normal cell adhesion contributes to enhanced
proliferation, migration, and invasion leading to metastasis. Fibronectin (FN)
is an extracellular matrix protein with putative roles in mediating these
actions. Indeed, tumor cells with decreased adhesiveness frequently fail to
deposit stromal FN (3). In particular, reduced FN expression has been noted in
transformed cell lines and primary tumors (4), including thyroid cancer (3, 5,
6), where diminished FN has been identified at the periphery of invasive tumor
margins. In this context, we found that down-regulation of FN stimulates
thyroid cancer cell proliferation and tumor growth (7). Conversely, 1,
25-dihydroxy vitamin D3 treatment increases cell adhesiveness and inhibits cell
proliferation and tumor growth through enhanced FN expression.
We will come back to fibronectin in out later analysis.
We have discussed E-cadherin at length in previous work. It
plays a critical role in stabilizing cell adhesion and localization. Loss of
E-cadherin results in loss of cell localization and thus cell movement.
Specifically in melanocytes the cells begin to leave the basal layer and
migrate upward as in melanoma in situ and downward as in superficial spreading
melanoma.
As Swiatoniowski et al state:
Integrins are molecules which play a significant role in
cell-extracellular matrix (ECM) interactions. They interact with the RGD
tripeptide of fibronectin (FN), one of the main components of ECM. Labile
expression of FN has been proven to play an important role both in the normal
developmental process (morphogenetic movements) and in the course of
carcinogenesis … Many authors have implicated loss or decrease of EC expression
as an independent negative prognostic marker in breast cancer patients (6-9).
There is increasing experimental evidence for a relationship between the EC
level and different features of breast cancer, including histological grade (7,
16) and axillary lymph node involvement (13-16)…. In conclusion, our experiment
revealed no prognostic value for EC or FN expressions in a homogenous group of
patients
Proteoglycans are single polypeptide with multiple sugars
attached. They provide for hydration in the ECM.
The proteases are ECM proteins which function to degrade the
refuse in the ECM. The metalloproteinases are a family of proteases. They are
also called MMP. MMP-9 and MMP-2 are ones of the MMPs often associated with
melanoma.
There has been extensive work examining the MMPs and
melanoma some dating back to the 1990s, see that of Luca et al. A recent result
by Hoffman et al state:
Matrix metalloproteinases (MMPs) and their tissue
inhibitors (TIMPs) are involved in tumour progression and metastasis. In this
study, we investigated the in vitro and in vivo expression patterns of MMP-1,
MMP-2, MMP-3, MMP-9, TIMP-1 and TIMP-2 mRNA and protein in a previously
described human melanoma xenograft model. This model consists of eight human
melanoma cell lines with different metastatic behaviour after subcutaneous
(s.c.) injection into nude mice. MMP-1 mRNA was detectable in all cell lines by
reverse transcription polymerase chain reaction (RT-PCR), but the expression
was too low to be detected by Northern blot analysis. No MMP-1 protein could be
found using Western blotting. MMP-2 mRNA and protein were present in all cell
lines, with the highest expression of both latent and active MMP-2 in the
highest metastatic cell lines MV3 and BLM. MMP-3 mRNA was expressed in MV3 and
BLM, and in the non-metastatic cell line 530, whereas MMP-3 protein was
detectable only in MV3 and BLM.
None of the melanoma cell lines expressed MMP-9. TIMP-1
and TIMP-2 mRNA and protein, finally, were present in all cell lines. A
correlation between TIMP expression level and metastatic capacity of cell lines,
however, was lacking. MMP and TIMP mRNA and protein expression levels were also
studied in s.c. xenograft lesions derived from a selection of these cell lines.
RT-PCR analysis revealed that MMP-1 mRNA was present in MV3 and BLM xenografts,
and to a lesser extent in 530. Positive staining for MMP-1 protein was found in
xenograft lesions derived from both low and high metastatic cell lines,
indicating an in vivo up-regulation of MMP-1. MMP-2 mRNA was detectable only in
xenografts derived from the highly metastatic cell lines 1F6m, MV3 and BLM. In
agreement with the in vitro results, the highest levels of both latent and
activated MMP-2 protein were observed in MV3 and BLM xenografts.
With the exception of MMP-9 mRNA expression in 530
xenografts, MMP-3, MMP-9, and TIMP-1 mRNA and protein were not detectable in
any xenograft, indicating a down-regulated expression of MMP-3 and TIMP-1 in
vivo. TIMP-2 mRNA and protein were present in all xenografts; interestingly,
the strongest immunoreactivity of tumour cells was found at the border of
necrotic areas. Our study demonstrates that of all tested components of the
matrix metalloproteinase system, only expression of activated MMP-2 correlates
with increased malignancy in our melanoma xenograft model, corroborating an
important role of MMP-2 in human melanoma invasion and metastasis.
We shall see the impact of MMPs as we examine the pathways.
Integrins are for the most part the receptors for ECM
proteins. They are one of many such cell surface receptors. The integrins play
important roles in cell homeostasis and cell to cell communications.
Let us briefly examine the gene MDA-9 and its protein Mda-9
and what is known and how it has evolved. Now MDA-9 is located on (8q12).
As the NIH data base states:
The protein encoded by this gene was initially identified
as a molecule linking syndecan-mediated signaling to the cytoskeleton. The
syntenin protein contains tandemly repeated PDZ domains that bind the
cytoplasmic, C-terminal domains of a variety of transmembrane proteins. This
protein may also affect cytoskeletal-membrane organization, cell adhesion,
protein trafficking, and the activation of transcription factors.
The protein is primarily localized to membrane-associated
adherens junctions and focal adhesions but is also found at the endoplasmic
reticulum and nucleus. Alternative splicing results in multiple transcript
variants encoding different isoforms.
In the paper, Src kinase activation is mandatory for
MDA-9/syntenin-mediated activation of nuclear factor-κB, by H
Boukerche, H Aissaoui, C Prévost, H Hirbec, S K Das, Z-Z Su, D Sarkar and P B
Fisher, the author’s state:
The scaffolding postsynaptic density-95/disks
large/zonula occludens-1 (PDZ) domain-containing protein melanoma differentiation
associated gene-9 (MDA-9)/syntenin is a tandem PDZ protein overexpressed in
human melanoma, and breast and gastric cancer cells. MDA-9/syntenin affects
cancer cell motility and invasion through distinct biochemical and signaling
pathways, including focal adhesion kinase and p38 mitogen-activated protein
kinase (MAPK), resulting in activation of the nuclear factor (NF)-κB pathway.
MDA-9/syntenin also promotes melanoma metastasis by
activating c-Src, but how c-Src regulates NF-κB activation is unclear. Using a
human melanoma model, we document that MDA-9/syntenin–c-Src interactions are
positive regulators of NF-κB activation. Inhibition of c-Src by PP2 treatment,
by blocking c-Src or mda-9/syntenin expression with small interfering RNA, or
in c-Src (−/−) knockout cell lines, reduces NF-κB activation following
overexpression of mda-9/syntenin or c-Src.
Deletion or point mutations of the PDZ binding motif
preventing MDA-9/syntenin association with c-Src reveals that both PDZ domains,
with PDZ2 being the dominant module, are required for activating downstream
signaling pathways, including p38 MAPK and NF-κB. We also document that
MDA-9/syntenin–c-Src complexes functionally cooperate with NF-κB to promote
anchorage-independent growth, motility and invasion of melanoma cells. These
findings underscore PDZ domains of MDA-9/syntenin as promising potential
therapeutic targets for intervening in a decisive component of cancer
progression, namely, metastatic tumor spread….
(MDA-9 Acts as a PDZ domain-containing adapter protein.
In adherens junctions, it couples syndecans to cytoskeletal proteins or
signaling components. Seems to be required for the targeting of TGF-alpha to
the cell surface in the secretory pathway. By virtue of its association with a
large number of additional proteins, including class B ephrins, TGF-alpha,
phosphotyrosine phosphatase, neurofaschin, neurexin, schwannomin/merlin, IL-5
receptor, various glutamate receptor subtypes, and the syndecan family of
heparan sulfate proteoglycans, MDA9 has been implicated in diverse processes,
including protein trafficking, activation of the transcription factor SOX4,
cytoskeleton-membrane organization, and cell adhesion/migration….
(MDA-9) Its expression is induced by IFN-gamma in melanoma
cells. Is believed to be involved in cancer metastasis. In melanoma, it
promotes the metastatic phenotype by activating NFkB and focal adhesion kinase
(FAK), which promotes induction of matrix metalloproteinase (MMP) and then
migration and extracellular matrix invasion of melanoma cells. Syntenin is
overexpressed and promotes cell migration in metastatic human breast and
gastric cancer cell lines.
The gene product is also called by many other names,
specifically:
1.
MDA9
2.
MDA-9
3.
TGF alpha cytoplasmic domain
interacting protein18
4.
TACIP18
5.
SYCL
6.
Syntenin-1
7.
Syndecan binding protein 1
8.
SDCBP
9. Melanoma differentiation associated protein 9
From Das et al. we have the following modified figure
:
Das et al state regarding the above pathway model:
Schematic diagram for mda-9/syntenin mediated NFk B activation. Upon interaction with ECM
(fibronectin), MDA-9/syntenin activates the p38/MAPK by augmenting FAK
phosphorylation. This results in degradation of Ik Ba and movement
of p65 from the cytoplasm where interaction with p50 results in binding to
target genes (MT1-MMP) resulting in enhanced production of MT1-MMP, which
interacts with TIMP-2 activating pro-MMP-2 to produce active MMP-2. This
product then enhances cell motility, invasion, and cancer cell growth.
mda-9/Syntenin activates the NF-kB pathway.
The original Figure appears to be from Boukerche et al as
shown with some mods below:
Note the differences. First the original shows multiple
integrins and multiple FAK binding and in turn a binding of MDA-9 initiating
the p38 pathway. Also note the explicit presence of NF-κB and its result of
genes forcing mobility, invasion and metastasis. The authors state:
Hypothetical model of signal transduction pathways
coordinately regulated by MDA-9/syntenin through its interaction with c-Src.
MDA-9/ syntenin interaction with c-Src results in clustering of c-Src/FAK
signaling complexes at high concentrations on the plasma membrane. The
activated c-Src/FAK complexes activate the p38 MAPK/NF-κB pathways that
regulate expression of genes involved in migration and invasion and thus play a
crucial role in MDA-9/syntenin-mediated tumor progression.
The initiation of NF-κB is a significant factor since this
transcription factor is what appears to be the instigator of the metastatic
processes.
From Pecorino, p 220, we have again presented the details
(as modified)
:
The above graphic clearly demonstrates the movement of the
transcription factor into the nucleus, from a bound state with IkB to an
unbound and active state. The target genes indicated includes an MMP gene which
again goes into the ECM.
As Sarkar et al state:
Melanoma differentiation associated gene-9 (mda-9),
also known as syntenin, is a PDZ domain– containing adapter protein that is
involved in organization of protein complexes in the plasma membranes,
regulation of B-cell development, intracellular trafficking and cell-surface
targeting, synaptic transmission, and axonal outgrowth. Recent studies now
define a seminal role for mda-9/sytenin in cancer metastasis.
Thus, Sarkar who is part of Fisher’s Lab at Virginia, have
had a focus on Mda-9. They continue:
Adapter proteins play an essential role in modulating
signal transduction from the extracellular environment to the intracellular
milieu by virtue of their association with key regulatory molecules … mda-9
was originally cloned as a gene differentially expressed in human melanoma
cells reprogrammed to terminally differentiate by combination treatment with
IFN-h
and the protein kinase C activator mezerein … Analysis of the
subcellular distribution of mda-9/syntenin revealed its
localization at the areas of cell-cell contact in cells of epithelial origin in
colocalization with F-actin, syndecan-1, E-cadherin, h-catenin,
and a-catenin
(12). In fibroblasts, mda-9/ syntenin localizes to
focal adhesions and in stress fibers. Overexpression of mda-9/syntenin
in different cells induces the formation of plasma membrane structures,
including ruffles, lamellipodia, fine extensions, and neurite-like structures,
showing its role in regulating the structure and function of the plasma
membrane…
They continue:
The major characteristic of malignant tumor cells is
their ability to invade foreign tissues and form metastatic foci at distant
locations in the body. Such a process requires tumor cell attachment to various
matrix proteins, degradation of the extracellular matrix (ECM) mainly by matrix
metalloproteinases (MMP), followed by migration into the surrounding stroma by
tumor cells…A model of progression of melanoma suggests that it begins by
conversion of a normal melanocyte into a benign nevi, subsequent transformation
into a radial and then a vertical growth phase primary melanoma, and finally
evolution into a metastatic melanoma.
Finally Sarkar et al outline the overall set of functions
which MDA-9 is involved in. Specifically they state:
1. Interleukin-5 signaling. mda-9/syntenin interacts
with interleukin- 5 (IL-5) receptor a and the transcription factor Sox4,
thus mediating IL-5–induced Sox4 activation …
2. Cell-surface trafficking.
Although mda-9/syntenin is located predominantly in the
plasma membrane, it is also identified in the early secretory pathway such as
the endoplasmic reticulum, intermediate compartment, and cis-Golgi,
thus facilitating cellsurface trafficking of secreted molecules such as proTGF-a,
an epidermal growth factor receptor ligand…
3. mda-9/syntenin and ephrin signaling.
Ephrins and their cellsurface tyrosine kinase receptors are implicated
in controlling axon guidance and fasciculation
…
4. Mediation of cohesiveness of epidermal stem cells.
In the basal layer of interfollicular epidermis the stem cells are
clustered, a feature known as cohesiveness. These cells express high levels of
Notch ligand D1, which is important for maintaining cohesiveness …
5. Regulation of glutamate signaling.
The excitatory neurotransmitter glutamate interacts with its cognate
receptors and regulates postsynaptic excitatory currents. Glutamate receptors
interact with mda-9/syntenin,
…
6. Regulation of axon outgrowth.
Unc51.1 is a serine/threonine kinase that is important for neurite
extension/parallel fiber formation in cerebellar granule neurons. mda-9/syntenin
interacts with Unc51.1 and Rab5, a member of the Ras-like small GTPases that is
a marker of early endosomes and is essential for endocytic membrane fusion and
trafficking. …
Boukerche et al in 2005 stated:
Studies using an enhanced green fluorescent protein mda-9/
syntenin fusion protein showed that endogenous mda-9/syntenin
colocalized with the E-cadherin complex and syndecan-1 at adherens junctions as
well as with focal adhesions and stress fibers at cell-substratum contact in
fibroblastic and epithelial cells. These findings suggest that Mda-9/syntenin might promote cytoskeletal
organizational changes and intracellular signaling.
The organization of these dissimilar focal contacts is
complex but was shown not only to contain the appropriate integrin but also
cytoskeletal proteins (vinculin, talin, and a-actinin) as well as
several cytoplasmic protein tyrosine kinases, including members of the src
family and focal adhesion kinase (FAK). Despite extensive research documenting
an ability of mda-9/syntenin to form multivalent interactions, little is
known about the role of Mda-9/syntenin
in cancer development.
Boukerche et al (2008) state:
Prior studies confirm that Mda-9/syntenin stimulates motility through
pathways involving FAK, p38MAPK, and NF-κB, leading to secretion of MMP-2 (4,
9). However, despite these intriguing observations, it is not fully understood
how Mda-9/syntenin orchestrates
these signaling molecules to enhance cancer cell motility and metastasis. A
complex network of protein-protein interactions characterizes the structural
organization of focal adhesions, involving known signaling molecules that play
functional roles in various cellular activities and other less well-defined
pathways.
We presently show that Mda-9/syntenin interacts with c-Src through
its PDZ domain and activates the c-Src/FAK signaling pathway to maximize tumor
cell motility and anchorage-independent growth of melanoma cells. Mda-9/Syntenin levels directly correlate
with increased c-Src activity in a human melanoma model that closely mimics the
early events of metastasis in humans.
In 2010 Boukerche et al report (also in Fisher’s Lab):
MDA-9/syntenin affects cancer cell motility and invasion
through distinct biochemical and signaling pathways, including focal adhesion
kinase and p38 mitogen-activated protein kinase (MAPK), resulting in activation
of the nuclear factor (NF)-kappaB pathway.
MDA-9/syntenin also promotes melanoma metastasis by
activating c-Src, but how c-Src regulates NF-kappaB activation is unclear.
Using a human melanoma model, we document that MDA-9/syntenin-c-Src
interactions are positive regulators of NF-kappaB activation. Inhibition of
c-Src by PP2 treatment, by blocking c-Src or mda-9/syntenin expression with
small interfering RNA, or in c-Src (-/-) knockout cell lines, reduces NF-kappaB
activation following overexpression of mda-9/syntenin or c-Src.
Deletion or point mutations of the PDZ binding motif
preventing MDA-9/syntenin association with c-Src reveals that both PDZ domains,
with PDZ2 being the dominant module, are required for activating downstream
signaling pathways, including p38 MAPK and NF-kappaB. We also document that
MDA-9/syntenin-c-Src complexes functionally cooperate with NF-kappaB to promote
anchorage-independent growth, motility and invasion of melanoma cells.
These findings underscore PDZ domains of MDA-9/syntenin
as promising potential therapeutic targets for intervening in a decisive
component of cancer progression, namely, metastatic tumor spread.
This set of papers from the Fisher Lab present several
interesting connections between the ECM and the intra-cellular signaling paths.
We have had prior arguments that one can develop models for metastasis by
examining the cell as a target entity and then by modeling the environment,
both the ECM and surrounding cells as influences on the target cell. In this
work we can expand it to include ECM factors in some detail.
The suggested control of other pathway elements, beyond just
the B-RAF control that we now have may be proven productive. Notwithstanding it
does establish a research path that is based upon established cell dynamics.
1. Beekman, J., P. Coffer, The ins and outs of syntenin, a
multifunctional intracellular adaptor protein, Journal of Cell Science 121,
1349-1355 Published by The Company of Biologists 2008.
2. Boukerche H., et al., Src kinase activation is
mandatory for MDA-9/syntenin-mediated activation of nuclear factor-κB, Oncogene.
29(21):3054-66, 2010 May 27.
3. Boukerche, H. et al, mda-9/Syntenin: A Positive Regulator of
Melanoma Metastasis, Cancer Res 2005; 65:10901-10911. Published online December
1, 2005
4. Boukerche, H. et al, mda-9/Syntenin promotes metastasis in human
melanoma cells by activating c-Src, pp 15914–15919, PNAS, October 14, 2008, vol. 105, no. 41.
5.
Cassimeris, L., et al,
Lewin’s Cell, 2nd Ed, Jones and Bartlett (Boston) 2011.
6. Das S., et al, MDA-9/syntenin: a positive gatekeeper of melanoma
metastasis, Frontiers in Bioscience 17, 1-15, January 1, 2012.
7.
Das, S., et al, Therapeutics,
Targets, and Chemical Biology Raf Kinase Inhibitor
RKIP Inhibits MDA-9/Syntenin-Mediated Metastasis in Melanoma, Cancer
Res Published Online First October 11, 2012.
8.
Hearing, V., S. Leong, From
Melanocytes to Melanoma, Humana (Totowa, NJ) 2006.
9. Ho, C., Stopping The Spread Of Melanoma By Removing Protein
Affecting Metastasis, RedOrbit, November 15, 2012.
10.
Hoffman, U., et al, Matrix
metalloproteinases in human melanoma cell lines and xenografts: increased
expression of activated matrix metalloproteinase-2 (MMP-2) correlates with
melanoma progression, British Journal of Cancer (1999) 81(5), 774–782.
11.
Houben, M., et al, Absence
of Classical MAP Kinase Pathway Signalling in Merkel Cell Carcinoma, Journal of
Investigative Dermatology (2006) 126, 1135–1142.
12. Hwangbo, C. et al, mda-9/Syntenin Protein Positively Regulates
the Activation of Akt Protein by Facilitating Integrin-linked Kinase Adaptor
Function during Adhesion to Type I Collagen, VOLUME 286 NUMBER 38 JOURNAL OF BIOLOGICAL CHEMISTRY, SEPTEMBER
23, 2011.
13.
Lacovara J., et al,
Fibronectin Enhancement of Directed Migration of B16 Melanoma Cells, Cancer
Research, 1984.
14.
Liu, W., et al, The
Melanoma-Associated Antigen A3 Mediates Fibronectin-Controlled Cancer
Progression and Metastasis, Cancer Res 2008;68:8104-8112. Published online
September 30, 2008.
15.
Luca, M., et al, Expression of Interleukin-8 by Human Melanoma Cells
Up-Regulates MMP-2 Activity and Increases Tumor Growth and Metastasis, American
Journal of Pathology, Vol. 151, No. 4, October 1997.
16. Pecorino, Molecular Biology of Cancer, Oxford (New York) 2nd
Ed, 2005.
17.
Ramos, D., et al, Analysis
of Integrin Receptors for Laminin and Type IV Collagen on MetastaticB16
Melanoma Cells, Cancer Research, 1990.
18. Sarkar, D., et al, mda-9/Syntenin: More than Just a Simple
Adapter Protein When It Comes to Cancer Metastasis, Cancer Res 2008; 68: (9).
May 1, 2008.
19. Swiatoniowski, G et al, E-cadherin and Fibronectin Expressions
Have No Prognostic Role in Stage II Ductal Breast Cancer, ANTICANCER RESEARCH
25: 2879-2884 (2005).
20.
Yi, M., E. Ruoslahti, A
fibronectin fragment inhibits tumor growth,
angiogenesis, and metastasis, pp 620–624, PNAS, January 16, 2001, vol.
98, no. 2.
21.
Zent, R., A., Pozzi,
Cell-Extracellular Matrix Interactions in Cancer, Springer (New York) 2010.