SPOP is part of the Hedgehog signalling pathway[1]. The Hedgehog signalling
pathway controls amongst other factors the formation of body segments in
insects and in vertebrates the development of the neural tube, limbs and
left-right asymmetry. In adult tissues Hedgehog is responsible for homeostasis,
equilibrium between cells loss and gain while maintaining total mass and
function. With an overactive Hedgehog pathway one sees excess cell proliferation
and tumor growth[2].
We demonstrate that below:

Thus SPOP has a controlling mechanism for cell replication.
Here Hedgehog attaches to Patched and the Patched inhibition of Smothered is
eliminated allowing Smothered to start a transcription process enabling
replication.
Now upon the activation of Smothered a set of processes are
activated and one product is a protein called the zinc finger transcription
factor Gli, which when mutually supported by SPOP allows movement to the
nucleus as a transcription factor activating the DNA to transcribe[3]. We depict that below:

From Barbieri et al we have the following putative relationships:
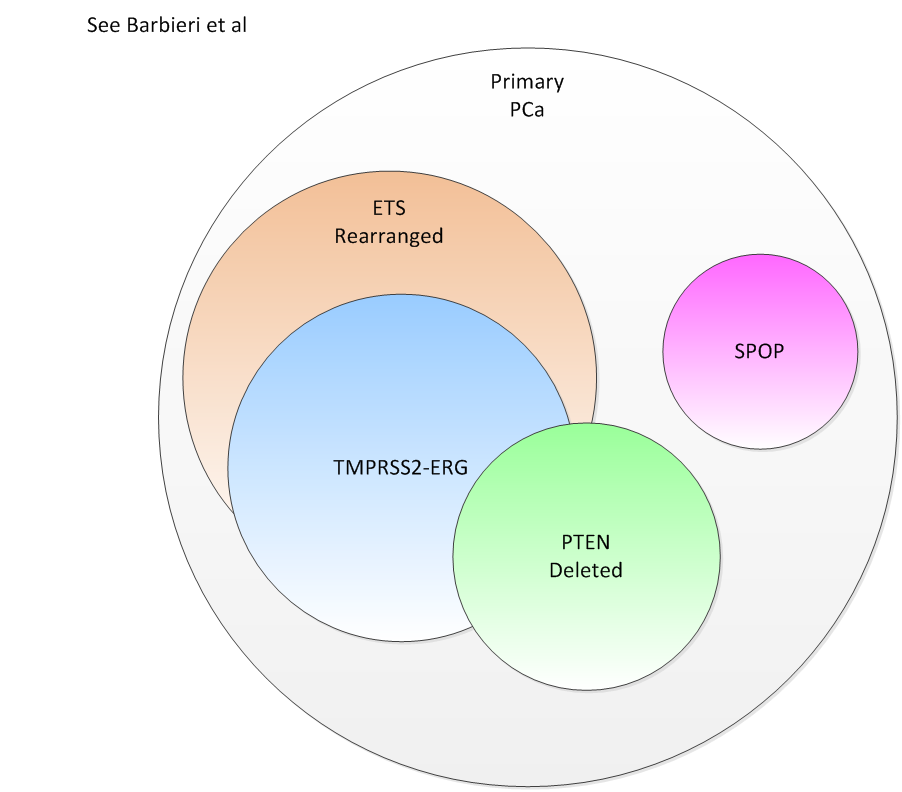
The authors argue that SPOP is a separate and significant
marker for PCa. The pathway involved is somewhat understood and is a
transcription driven pathway initiated by Hedgehog activation and Patched
suppression with Smothered activation. From the NCI pathway databases we have a
putative requirement that SPOP is needed to activate GLI for subsequent
transcription and cell reproduction.
Specifically Barbieri et al state:
As demonstrated by a subsequent analysis of significantly
more genomes, there are only a few truly recurrent non-synonymous mutations in
PCa. The most common
recurrent non-synonymous mutation in PCa involves SPOP.
The SPOP gene encodes for the substrate-recognition component of
a Cullin3-based E3-ubiquitin ligase. Mutations
in SPOP in PCa were reported originally in two systematic
sequencing studies. We have
now identified the presence of recurrent mutations in SPOP in
6–13% of human PCas in multiple independent patient cohorts.
Recurrent missense mutations are found exclusively in the structurally defined substrate-binding cleft of SPOP, and structural analysis suggests that these mutations will inactivate SPOP function by disrupting SPOP–substrate interaction.
Further, we found that loss of SPOP function in prostate cell lines resulted in increased invasion and altered gene expression; evidence of this expression signature was identified in primary tumours harbouring SPOP mutation.
Importantly, all SPOP mutations occurred in tumours that were negative for ERG rearrangement; these tumours displayed characteristic somatic copy number aberrations. Taken together, these findings support a distinct molecular class of PCa.
Recurrent missense mutations are found exclusively in the structurally defined substrate-binding cleft of SPOP, and structural analysis suggests that these mutations will inactivate SPOP function by disrupting SPOP–substrate interaction.
Further, we found that loss of SPOP function in prostate cell lines resulted in increased invasion and altered gene expression; evidence of this expression signature was identified in primary tumours harbouring SPOP mutation.
Importantly, all SPOP mutations occurred in tumours that were negative for ERG rearrangement; these tumours displayed characteristic somatic copy number aberrations. Taken together, these findings support a distinct molecular class of PCa.
In a recent Nature Medicine article the same authors relate[4]:
Prostate cancer is the second most common cancer in men
worldwide and causes over 250,000 deaths each year. Overtreatment of indolent
disease also results in significant morbidity. Common genetic alterations in
prostate cancer include losses of NKX3.1 (8p21) and PTEN (10q23), gains of AR
(the androgen receptor gene) and fusion of ETS family transcription factor
genes with androgen-responsive promoters.
Recurrent somatic base-pair substitutions are believed to be less contributory in prostate tumorigenesis but have not been systematically analyzed in large cohorts. Here, we sequenced the exomes of 112 prostate tumor and normal tissue pairs. New recurrent mutations were identified in multiple genes, including MED12 and FOXA1. SPOP was the most frequently mutated gene, with mutations involving the SPOP substrate-binding cleft in 6–15% of tumors across multiple independent cohorts.
Prostate cancers with mutant SPOP lacked ETS family gene rearrangements and showed a distinct pattern of genomic alterations. Thus, SPOP mutations may define a new molecular subtype of prostate cancer.
Recurrent somatic base-pair substitutions are believed to be less contributory in prostate tumorigenesis but have not been systematically analyzed in large cohorts. Here, we sequenced the exomes of 112 prostate tumor and normal tissue pairs. New recurrent mutations were identified in multiple genes, including MED12 and FOXA1. SPOP was the most frequently mutated gene, with mutations involving the SPOP substrate-binding cleft in 6–15% of tumors across multiple independent cohorts.
Prostate cancers with mutant SPOP lacked ETS family gene rearrangements and showed a distinct pattern of genomic alterations. Thus, SPOP mutations may define a new molecular subtype of prostate cancer.
This just adds another gene in the mix for PCa. Namely they
authors argue that it is a different type. We would still ask the same
questions:
1. What is the issue regarding the presence or absence of a
CSC stem cell in PCa.
2. When does this mutation occur.
3. What causes the mutation.
4. SPOP is not a true kinase so what type of blocking would
be possible to mitigate the presence of a mutant.
References
1. Barbieri, C. et al, Molecular genetics of prostate cancer:
emerging appreciation of genetic complexity, Histopathology 2012, 60, 187–198.
2.
Barbieri, C., et al, Exome
Sequencing Identifies Recurrent SPOP, FOXA1 and MED12 Mutations in Prostate
Cancer, Nature Genetics (2012).
3.
Marks, F., et al, Cellular
Signal Processing, Garland (New York) 2009.
4.
Pecorino, L, Molecular
Biology of Cancer, Oxford (New York) 2008.
[1] http://pid.nci.nih.gov/search/MoleculePage?molid=203488 and http://pid.nci.nih.gov/search/search_landing.shtml?atom_id=208460,208462&what=graphic&jpg=on
and pathway at http://pid.nci.nih.gov/search/advanced_landing.shtml?what=graphic&svg=&jpg=true&xml=&biopax=&complex_uses=on&family_uses=on°ree=1&molecule=&pathway=hedgehog¯o_process=&source_id=5&evidence_code=NIL&evidence_code=IAE&evidence_code=IC&evidence_code=IDA&evidence_code=IFC&evidence_code=IGI&evidence_code=IMP&evidence_code=IOS&evidence_code=IPI&evidence_code=RCA&evidence_code=RGE&evidence_code=TAS&output-format=graphic&Submit=Go
[2]
See Marks et al p 210-212.
[3]
See Pecorino, p. 168-170.


