The focus on telomeres and cancer has been an area of active
interest for almost two decades. As Shay et al (2012) state:
To grow indefinitely, human cancer cells must counteract
the progressive loss of telomeric DNA that universally accompanies cell
division. To do this, about 85 to 90% of cancers use telomerase, an enzyme that
synthesizes the tandem 5′-TTAGGG-3′ hexanucleotide repeats of telomeric DNA by reverse
transcription using its own RNA subunit as a template. Because telomerase is
not expressed in most normal human cells, telomerase inhibition is considered
an almost universal oncology target, and several clinical trials are under way
The above focuses on the critical importance of telomerase.
Before continuing it is worth reviewing the telomere. As Shay and Wright state:
Telomeres are tracts of repetitive DNA (TTAGGG/ AATCCC
for human telomeres) that protect chromosomes from degradation and loss of
essential genes, and allow the cell to distinguish between double-strand breaks
and natural chromosome ends. Human telomeres at birth contain 15–20-kilobase
pairs of the repetitive sequence TTAGGG followed
by a 3′ single-strand
overhang on the G-rich strand, which is believed to be inserted within the
double-stranded region to give a lariat-like structure called a t-loop.
Telomeres progressively shorten in most human cells with
increased age, and telomere length in almost all middle-aged human tissues is
approximately half that of the new born length. Telomere-specific proteins
(such as protection of telomeres-1 (POT1), telomeric repeat-binding factor-1 (TRF1)
and TRF2) bind directly to the single- and double-strand telomere regions to
form a complex, providing a cap over the ends of the chromosomes that protects
chromosome termini from degradation, recombination and end-joining reactions.
The authors further state that telomeres are somewhat
maintained in humans via the use of telomerase as follows:
Telomere length is maintained by a balance between
processes that lengthen telomeres, such as the activity of the cellular
ribonucleoprotein enzyme complex telomerase, and processes that shorten
telomeres, such as incomplete synthesis of the lagging DNA strand and end processing
events. Telomerase stabilizes telomere length by adding TTAGGG repeats onto the
telomeric ends of the chromosomes, thereby compensating for the continued
erosion of telomeres that occurs in its absence. Human telomerase contains two essential
components, a telomerase reverse transcriptase catalytic subunit (hTERT) and a functional telomerase RNA
(hTR, also known as TERC…
Other earlier authors such as Campisi et al state:
Telomeres are the repetitive DNA sequences and
specialized proteins that form the distinctive structure that caps the ends of
linear chromosomes. Telomeres allow cells to distinguish the chromosome ends
from double strand DNA breaks. The telomeric structure prevents the degradation
or fusion of chromosome ends, and thus is essential for maintaining the
integrity and stability of eukaryotic genomes. In addition and perhaps less
widely appreciated, telomeres may also indirectly influence gene expression.
The length, structure and organization of telomeres
are regulated by a host of telomere-associated proteins, and can be influenced
by basic cellular processes such as cell proliferation, differentiation, and
DNA damage. In mammalian cells, telomere length and/or telomere structure have
been linked to both cancer and aging. Here, we briefly review what is known
about mammalian telomeres and the proteins that associate with them, and
discuss the cellular and organismal consequences of telomere dysfunction and
the evidence that cells with dysfunctional telomeres can contribute to cancer
and aging phenotypes.
As reported in the Harvard Gazette we have[1]:
Two mutations that collectively occur in 71 percent of
malignant melanoma tumors have been discovered in what scientists call the
“dark matter” of the cancer genome, where cancer-related mutations haven’t been
previously found….
This non-coding DNA, much of which was previously
dismissed as “junk,” accounts for 99 percent of a cell’s genome. A large number
of oncogenic mutations in cancer have been identified in the past several
decades, but all have been found within the actual genetic blueprints for
proteins….
“In addition, this represents the discovery of two of the most prevalent
melanoma gene mutations. Considered as a whole, these two TERT
promoter mutations are even more common than BRAF
mutations in melanoma. Altogether, this discovery could cause us to
think more creatively about the possible benefits of targeting TERT in cancer
treatment or prevention,” Garraway said.
The mutations affect a promoter region — a stretch of DNA
code that regulates the expression of a gene — adjacent to the TERT gene. TERT
contains the recipe for making telomerase reverse transcriptase, an enzyme that
can make cells virtually immortal, and is often found overexpressed in cancer
cells. A promoter region of DNA controls the rate of a gene’s transcription
— the copying of its DNA recipe into a message used by the cell to manufacture
a protein….
The researchers said the same mutations are present in
cell lines from some other malignancies, and that preliminary evidence showed
they might be unusually common in bladder and liver cancers. They also noted
that the discovery of these important mutations in DNA previously not linked to
cancer-causing alterations highlights the value of whole-genome searches of
tumor DNA.
Another report on Science 2.0 states[2]:
They analyzed the genomes of family members and found an
identical mutation in the gene for telomerase, an enzyme often called
'immortality enzyme', in all persons studied. Telomerase protects the ends of
chromosomes from being lost in the process of cell division and, thus, prevents
that the cell ages and dies. The inherited gene mutation leads to the formation
of a binding site for protein factors in the controlling region of the
telomerase gene, causing it to become overactive. As a result, mutated cells
overproduce telomerase and hence become virtually immortal.
This finding prompted the scientists to also look for
mutated telomerase genes in non-inherited (sporadic) melanoma, which is much
more common than the familial variant. In most of the tissue samples of
melanomas of all stages they found alterations in the telomerase gene switch,
which the researchers clearly identified as typical consequences of sun
exposure. Even though these mutations were not identical to those found in the
melanoma family, they had the same effect: overactive telomerase…
This is also confirmed by the surprising incidence of
this alteration: The telomerase gene is the most frequently mutated gene in
melanoma. "This is something we hadn't expected, because malignant
melanoma has been genetically analyzed thoroughly. But this mutation always
seems to have been overlooked," says Kumar.
It should be noted in the above the reference to sun
exposure. The argument is that the telomerase change is a direct consequence of
the UV exposure. We will focus on that observation later. The “overlooked”
nature of this gene and its product is also of issue in that many researchers
have examined telomerase extensively so frankly it is not truly new, even as a target
for control.
Before continuing it is worth a quick summary of TERT, the
telomerase that maintains the telomere. TERT is located at 5p15.33. From NCBI
we have[3]:
Telomerase is a ribonucleoprotein polymerase that
maintains telomere ends by addition of the telomere repeat TTAGGG. The enzyme
consists of a protein component with reverse transcriptase activity, encoded by
this gene, and an RNA component which serves as a template for the telomere
repeat. Telomerase expression plays a role in cellular senescence, as it is
normally repressed in postnatal somatic cells resulting in progressive
shortening of telomeres. Deregulation of telomerase expression in somatic cells
may be involved in oncogenesis.
Studies in mouse suggest that telomerase also participates
in chromosomal repair, since de novo synthesis of telomere repeats may occur at
double-stranded breaks. Alternatively spliced variants encoding different
isoforms of telomerase reverse transcriptase have been identified; the
full-length sequence of some variants has not been determined. Alternative
splicing at this locus is thought to be one mechanism of regulation of
telomerase activity.
The observation can be made that if we do not have adequate
TERT then the Telomere ends decay and ultimately the cell line dies off. This
is the typical case. Therefore take a malignant melanoma cell. If it has in its
pathways and receptors been activated to mitotic duplication then if the TERT
is inadequate then the Telomere ends get cut shorter each time it goes through
mitosis and at some point it just stops. For example, and this is just for
exemplar purposes only, we have a malignant melanocyte, then it goes through
mitosis say 10,000 times but each time it would lose a piece of the Telomere
until they are all gone, then th cell cannot go again. But if there is an
overabundance of TERT, then the TERT resupplies what may be lost and this cell
has no way of stopping, at least due to this factor.
The ETS family of genes is positive or negative regulators
of gene expression. They can up or down regulate expression. They are named for
the initial gene discovered, the E26 Transforming Sequence, where E26 was the
oncogene v-ets characterized in 1986 of an avian transforming virus called E26.
It is also called the erythroblast transforming specific family, as discussed
by Zong et al.
The ETS family is a large family of over 20 such genes, and
we will focus on ERG specifically. The Table below is from Watson et al.
Subgroup
|
Name
|
Unigene Name
|
Alternative
Names
|
Locus
|
Size
|
|
1
|
ETS
|
ETS1
|
ETS1
|
11q23.3
|
441
|
|
2
|
ETS2
|
ETS2
|
21q22.3
|
469
|
||
3
|
ERG
|
ERG2
|
ERG
|
21q22.3
|
462
|
|
4
|
FLI1
|
FLI1
|
ERGB
|
11q24.1-q24.3
|
452
|
|
5
|
FEV
|
FEV
|
2q36
|
238
|
||
6
|
PEA3
|
PEA3
|
ETV4
|
E1AF, PEAS3
|
17q21
|
462
|
7
|
ERM
|
ETV5
|
3q28
|
510
|
||
8
|
ER81
|
ETV1
|
7p21.3
|
458
|
||
9
|
ETV
|
ER71
|
ETV2
|
ETSRP71
|
19q13.12
|
370
|
10
|
TCF
|
ELK1
|
ELK1
|
Xp11.2
|
428
|
|
11
|
SAP1
|
ELK4
|
1q32
|
431
|
||
12
|
NET
|
ELK3
|
SAP2, ERP
|
12q23
|
407
|
|
13
|
GABP
|
GABP α
|
GABPA
|
E4TF1
|
21q21.3
|
454
|
14
|
ELF1
|
ELF1
|
ELF1
|
13q13
|
619
|
|
15
|
NERF
|
ELF2
|
NERF1, NERF2, EU32
|
4q28
|
581
|
|
16
|
MEF
|
ELF4
|
ELFR
|
Xq26
|
663
|
|
17
|
SPI1
|
SPI1
|
SPI1
|
PU.1, SFPI1, SPI-A
|
11p11.2
|
264
|
18
|
SPIB
|
SPIB
|
19q13.3-q13.4
|
262
|
||
19
|
SPIC
|
SPIC
|
12q23.2
|
248
|
||
20
|
TEL
|
TEL
|
ETV6
|
12p13
|
452
|
|
21
|
TEL2
|
ETV7
|
TEL-B
|
6p21
|
264
|
|
22
|
ERF
|
ERF
|
ERF
|
19q13
|
548
|
|
23
|
PE-1
|
ETV3
|
METS
|
1q21-q23
|
250
|
|
24
|
PDEF
|
PDEF
|
SPDEF
|
6p21.3
|
335
|
|
25
|
ESE
|
ESE1
|
ELF3
|
ESX, JEN, ERT,
EPR1
|
1q32.2
|
371
|
26
|
ESE2
|
ELF5
|
11p13-p12
|
255
|
||
27
|
ESE3
|
EHF
|
ESEJ
|
11p12
|
300
|
The ERG gene was first presented in the paper by Shyam and
Reddy et al in 1987. There the authors identified it and set it in the ETS
family. From Weinberg, we see that the ETS are transcription factors driven by
the RAS/RAF pathway along with other such factors.

ETS also plays a significant role in the process. We briefly
review that as well. ETS is located at 11q23.3. From NCBI we have[4]:
This gene encodes a member of the ETS family of
transcription factors, which are defined by the presence of a conserved ETS
DNA-binding domain that recognizes the core consensus DNA sequence GGAA/T in
target genes. These proteins function either as transcriptional activators or
repressors of numerous genes, and are involved in stem cell development, cell
senescence and death, and tumorigenesis. Alternatively spliced transcript
variants encoding different isoforms have been described for this gene
From Smalley and Flaherty we have the following pathway for
ETS:

The mutations we discuss here are somewhat new and they are
present in a relatively large number of samples, at least percentage wise. We
know that ETS has transcription control and we can see from above the
relationship to BRAF as well. Thus there are many points of loss of control in
a melanoma cell. Specifically, as Chudnovsky et al note[5]:
Multiple genetic alterations occur in melanoma, a lethal
skin malignancy of increasing incidence. These include mutations that activate
Ras and two of its effector cascades, Raf and phosphoinositide 3-kinase (PI3K).
Induction of Ras and Raf can be caused by active N-Ras and B-Raf mutants as
well as by gene amplification. Activation of PI3K pathway components occurs by
PTEN loss and by AKT3 amplification. Melanomas also commonly show impairment of
the p16(INK4A)-CDK4-Rb and ARF-HDM2-p53 tumor suppressor pathways. CDKN2A
mutations can produce p16(INK4A) and ARF protein loss. Rb bypass can also occur
through activating CDK4 mutations as well as by CDK4 amplification. In addition
to ARF deletion, p53 pathway disruption can result from dominant negative TP53
mutations. TERT amplification also occurs in melanoma. The extent to which these
mutations can induce human melanocytic neoplasia is unknown. Here we
characterize pathways sufficient to generate human melanocytic neoplasia and
show that genetically altered human tissue facilitates functional analysis of
mutations observed in human tumors.
As Horn et al state:
Cutaneous melanoma occurs in both familial and sporadic
forms. We investigated a melanoma-prone family through linkage analysis and
high-throughput sequencing and identified a disease-segregating germ line
mutation in the promoter of the telomerase reverse transcriptase (TERT) gene,
which encodes the catalytic subunit of telomerase. The mutation creates a new
binding motif for Ets/TCF transcription factors near the transcription start
and in reporter gene assays, caused up to 2-fold increase in transcription. We
then screened the TERT promoter in sporadic melanoma and observed recurrent UV
signature somatic mutations in 125/168 (74%) of human cell lines derived from
metastatic melanomas, corresponding metastatic tumor tissues (45/53, 85%) and
in 25/77 (33%) primary melanomas. The majority of those mutations occurred at
two positions in the TERT promoter and also generated binding motifs for
ETS/TCF transcription factors.
Horn et al conjecture the following pathway:

As Huang et al state:
Systematic
sequencing of human cancer genomes has identified many recurrent mutations in
the protein coding regions of genes but rarely in gene regulatory regions. Here
we describe two independent mutations within the core promoter of TERT, the
gene coding for the catalytic subunit of telomerase, which collectively occur
in 50 of 70 (71%) of melanomas examined. These mutations generate de novo
consensus binding motifs for ETS transcription factors, and in reporter assays
the mutations increased transcriptional activity from the TERT promoter by 2 to
4-fold. Examination of 150 cancer cell lines derived from diverse tumor types
revealed the same mutations in 24 cases (16%), with preliminary evidence of
elevated frequency in bladder and hepatocellular cancer cells. Thus, somatic
mutations in regulatory regions of the genome may represent an important
tumorigenic mechanism.
We have discussed before the Wnt pathway connection to TERT
as well. As shown below we have discussed this option as well.

This has been discussed by Hoffmeyer as well as by Greider.
As Greider states:
Recent studies have proposed that the Wnt pathway is
linked to TERT in a quite different way. Constitutive overexpression of TERT in
mice activates the Wnt pathway, suggesting that TERT may also function as a
transcription factor. Although one study did not observe Wnt pathway activation
in response to TERT overexpression, other studies have raised questions about
the physiological relevance of the constitutive overexpression of TERT.
Deletion of TERT in mice does not affect expression of target genes in the Wnt
pathway, nor give rise to the cellular phenotypes that loss of Wnt signaling
induces, indicating that TERT regulation of Wnt signaling may be limited to situations
where TERT is overexpressed.
It is reasonable to propose that Wnt regulates TERT given that Wnt signaling plays an essential
role in stem cell self-renewal and that TERT is needed for the long-term growth
of stem cells. TERT regulation seems to require not one, but two master
transcriptional regulators to assure that there is neither too much, which may
allow the growth of cancer cells, nor too little, which might lead to stem cell
failure. The finding by Hoffmeyer et al. that both β-catenin
and Klf4 are required to activate TERT expression puts the horse
(Wnt) before the cart (TERT) and provides a foundation for linking telomerase
levels and self-renewal.
Thus TERT regulation is truly a complex process. We have
examined the impact of Wnt on melanoma previously. This recent work is on
mutations on TERT genes yet we also must consider the influence of Wnt as well.
Observations
This discovery leads to several observations of note:
1. One could have imagined something of this happening with
Telomeres. It would almost be necessary to allow ongoing uncontrolled mitotic
activity. Thus, despite the fact that there is no surprise here we do have a
specific target, namely the activator of TERT.
2. Melanoma, as most other cancers, has a multiplicity of
changes to genes. There are ligands, receptors, pathway elements, transcription
factors, and the telomere issues as well. It is clear that no single factor is
the dominant one as of yet. BRAF as a target works for a while and then there
is a work around. Thus cancer is an evolving process, and one which may be
highly adaptive.
3. A Conjecture: As we have learned more and more as to
aberrant genes and their products, as well as miRNAs, and their effects, one
could envision several uses of malignancy profiling. We consider that in two
steps.
Step 1: Profiling a Specific Patient at Various Locations.
As shown below we consider a specific patient and then profile gene expression
as a function of distance from the site of initiation, if such was possible.
Then we can see how various aberrant genes are being expressed over the
distances measure from the source. One would suspect that distance must be
measured in some normalized manner but we leave that as an exercise for the
student at this time. This gives us a profile for a specific patient, perhaps
one for developing therapeutics.

Step 2: The Same Location but across a Large Pool of
Patients: Again we look now at the same distance from the source, perhaps at
the same time, again an exercise for the student, and we get profiles of the
expression of aberrant genes. This allows us to understand the between patient differences.
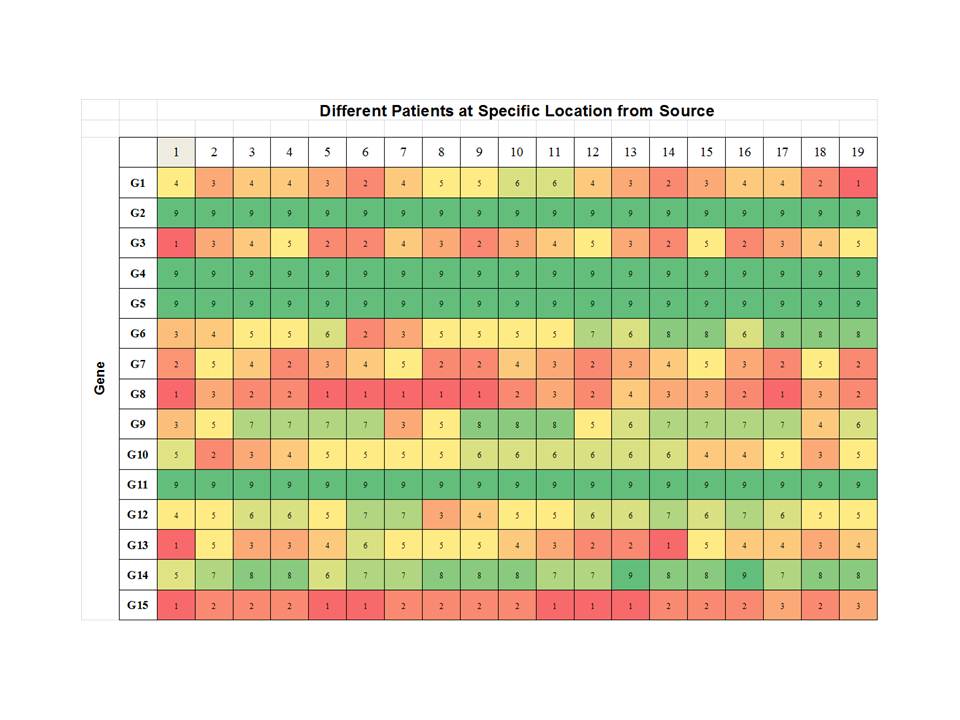
3. Is Seventy Enough? The study did an analysis on 70
lesions. Perhaps that is not enough. Furthermore based upon our previous
comments perhaps a correlative study is demanded as well, by patient and by
distance.
4. One of the problems I see is the continually hyping of
the results as if this is finally the right answer. Anyone even slightly
familiar with the field will understand that each input is vital but assembling
them in a cohesive whole is essential. The systems approach is the sine qua
non, but that cannot be done without the continual bench work required to
understand the details.
For example in an article in the Boston Globe the reporter
states[6]:
Now scientists working independently in Boston and
Germany have made a surprising discovery: a set of genetic mutations found in
most melanomas, the deadliest skin cancer. The presence of these mutations in
the vast majority of tumors studied suggests that the researchers may have
stumbled upon a fundamental mechanism involved in a hallmark trait of cancer
cells—their ability to live forever—that could one day be targeted by drugs.
Outside researchers said the work, published online
Thursday in the journal Science Express, is exciting because the conclusion is
the opposite of what many exhaustive studies of cancers have shown.
In reality as we have discussed, it was imperative that the
Telomeres be preserved in metastasis. Millions of rapid mitotic changes in a
stem cell must survive and that means keeping Telomeres and that means lots of
TERT. Somehow the conclusion was logical, consistent and not at all unexpected
especially given what else has been found in the past decade.
The article continues:
Both teams zeroed in on mutations in a part of the genome
called a promoter, which acts like a volume knob on a stereo to control gene
activity. The gene that the promoter controlled happened to be one that has
long been of interest in cancer because it creates part of an enzyme called
telomerase, which enables cancer cells to continue to divide indefinitely as
one of its key jobs. Still, it wasn’t easy for the researchers to convince
themselves that what they found, underlying more than two-thirds of melanoma
cases, was real.
One would expect this and if one looks at say the miRNA
discoveries, they all add up to what controls the ultimate expression of
mitotic survival.
5. Theraputics: Can we expect therapeutics from this
understanding? Good question. Kinase inhibitors are now well understood, one
could in theory build an inhibitor here as well. Is this the target, another
target, necessary, helpful, we can only guess. Yet the above Conjecture may
allow for the development of a therapeutic profiling plan for melanoma and
other malignancies.
References
1.
Campisi, J., et al,
Cellular senescence, cancer and aging: the telomere connection, Exp Geront V 36
pp 1619-1637, 2001.
2.
Chudnovsky, Y., et al, Use
of human tissue to assess the oncogenic activity of melanoma-associated
mutations, Nat Genet. 2005 Jul;37(7):745-9. Epub 2005 Jun 12.
3.
Greider, C., Wnt Regulates
TERT – Putting the Horse Before the Cart, Science, V 336 p 1519, 2012.
4.
Hoffmyer, K., et al,
Wntβ-Catenin Signalling Regulates Telomeres in Stem Cells and Cancer Cells,
Science, V 336 p 1549, 2012.
5.
Horn, S. e al, TERT
Promoter Mutations in Familial and Sporadic Melanoma, Sciencexpress, 24 January
2013.
6.
Huang, F., et al, Highly
Recurrent TERT Promoter Mutations in Human Melanoma, Sciencexpress, January
2013.
7.
Ip, J., Telomeres and
Cancer: A New Approach to Therapy, Bio Teach Jrl, V 2 Fall 2004.
8.
Neumann, A., R. Reddel,
Telomere Maintenance and Cancer – Look, no Telomerase, Nature Review Cancer, V
2, Nov 2002.
9.
Prescott, J., et al,
Epidemologic Evidence for the Role of Telomere Dysfunction in Cancer Etiology,
Mutation Res, 2011.
10.
Shay, J. et al, Cancer and
Telomeres - An ALTernative to Telomerase, Science, 15 June 2012, Vol 336, pp
1388-1390.
11.
Shay, J., W. Wright, Telomerase therapeutics for cancer: challenges and
new directions, Nature Reviews Drug Discovery AOP, published online 9 June
2006.
12.
Shyam, E., et al, The erg gene: A human gene related to the ets
oncogene, Proc Nat Acad Sci, Sept 1987.
13. Smalley K., K. Flaherty,
Integrating BRAF/MEK inhibitors into combination therapy for melanoma, Brit Jrl
Can 2007.
14. Watson, J., et al, Molecular Biology of the Gene,
Benjamin Cummings (San Francisco) 2004.
15. Weinberg, R., The
Biology of Cancer, Garland (New York) 2007.
16. Zong, Y., et al, ETS
Family Transcription Factors Collaborate with Alternative Signalling Pathways
to Induce Carcinomas from Adult Murine Prostate Cells, PNAS, V 106, 209, pp
12465-12470.


