There has recently been several pathway control mechanisms developed and tested and also an immunological approach deemed to be somewhat effective. We examine them here and also use them as suggestive of what else may be accomplished.
The current methods focus on two areas: (i) controlling aberrant pathways and (ii) using the immune response to control aberrant cells. It should be noted that in both cases we are dealing with the paradigm of a single but multiplying yet identical cancer cell. There is no hypothesis as regards to a stem cell or to the fact that the cancer may be multi-clonal.
1.1.1 Aberrant Pathway Control
We now examine the aberrant pathway approach. First let us consider the pathways that control a single cell. We show them below:

The above shows two results; cell proliferation and cell survival. They are two characteristics of a cancer. Namely the cell replicates and it does so in an almost immortal manner. The changed cell then starts to take over where other functional cells have been and the result is an unstable and ultimately deadly takeover of the human. Thus the two pathways are but a few of the many we will discuss at length. Yet the key point is that in examining melanoma it has been discovered that there is a specific mutation in the B-RAF gene that activate the MEK pathway. Activating that pathway creates a situation where we have an uncontrolled growth.
The growth factors activate the RTK kinase which activates the RAS which activates a B-RAF which overexpresses its product and this over-expression is what drives the proliferation pathway. It is this single gene and its protein expression which causes the problem in 60% of the cases.
The cell survival is often controlled by PTEN and it is the loss of PTEN which results in the cancer cell immortality. The PTEN loss is comparable to the same issue we have seen in prostate cancer.
Key to aberrant pathway control is a simple principle. Namely, we base the approach on the observed fact that certain pathway control elements have been changed as a result of a change in the underlying gene. We will show that in the current well known example of B-RAF that the underlying gene of B-RAF has been mutated and it the resulting B-RAF protein which has allowed the pathway to be turned on permanently. Thus the putative solution is to turn off the protein by targeting it with a drug which will pass the cell membrane and bind to the protein and inactivate it. A simple approach based upon an established fact. As we shall see there are two immediate issues: (i) only about 50% of the melanoma patients have the mutation, and (ii) the drug lasts for a relatively short time. It is similar to the effect that imatinib has on CML, a temporary regression and then a return.
As we shall see the possible solution may be multiple drug therapies targeting other pathway elements.
Now another way to view the pathways is shown below with the prominent role of c-Myc displayed at a common point. Note here we have the common surface kinases and the impact of B-RAF as well as PTEN. PTEN can modulate the limited up-regulation of B-RAF but only to a degree. As we have seen in PCa the loss of PTEN functionality leads to very aggressive forms.

The above also presents alternative control elements for possibly melanoma or frankly many other cancers. Specifically Smalley and Flaherty (2009) had suggested these pathway elements focusing on B-RAF, AKT and PI3K. One could also focus directly on the genes through a suppression mechanism but the technology for doing so is not yet available. Also there must be some specific targeting since we do not desire to target normal types of these products.
The control of aberrant pathways is conceptually simple.
1. Using a methodology such as microarrays, attempt to identify genes, or their expressions, which are present in the malignant cell. These are not unique and sometimes they are transient as well. The B-RAF identification is an example.
2. Develop a target molecule which can attach to and inactivate the aberrant gene or protein. In the current case of B-RAF they have deactivated the protein.
3. Test and use.
It may sound simple but the first step is potentially searching for a needle in a haystack and the second step can be as demanding. One may ask why not just block MEK or AKT just to stop everything. Assuming targets are possible the problem is it would do so for all cells and it would play havoc on the rest of the body. No blood cells, no hair, skin, and the like.
1.1.2 B-RAF control
The most recent one is the control of a mutated B-RAF, a variant of the RAF pathway. It was observed that there was a mutation in the B-RAF gene so that what was produced was a different B-RAF called V600E which had excessive up-regulation in almost 50-60% of all metastatic melanomas. The identification of this product then allowed for its targeting and suppression as a means to reduce cell proliferation. The results have been reported recently by the work of Chapman et al (2011) and Flaherty et al (2010). A review by Smalley and Flaherty (2009) had made suggestions on controlling both the BRAF as well as the AKT pathway. We will discuss that later. Recent work by Poulikakos and Solit (2011) has also presented both BRAF and MEK control, trying to avoid the loss of efficacy we discuss here.
Specifically, a drug now called Vemurafenib, or PLX4032, binds to the ATP activation site on the B-RAF mutation V600E and as such it blocks the overexpression of this protein and reduces the flow downward which we have shown causes ultimately an up-regulation of proliferation.

Now we can also see that Vemurafenib can lose its effectiveness and there are several proposals for why this happens. We discuss a few here. From Solit and Rosen (2011) we show one of the possible ways in which resistance can occur. We discuss several of their conjectures in detail.
Below we depict the supposition from Solit and Rosen. Arguably this is what accounts for the mortality in the Kaplan Meir data they have from their trials.

The paper by Solit and Rosen propose three reasons for loss of action of PLX4032:
(i) In melanomas with the BRAF V600E mutation, levels of activated RAS are too low to promote adequate formation of RAF dimers, and PLX4032 inhibits RAF activity and ERK signaling … This model is consistent with our observation that the introduction of mutant (activated) RAS into cells with mutant BRAF causes insensitivity of the ERK pathway to the drug. This model suggests that increases in RAF dimerization (because of RAS activation or increased RAF expression) will cause ERK signaling to become insensitive to PLX4032 …
(ii) The findings of Johannessen et al. suggest another mechanism for the resistance of ERK signaling to RAF inhibition in cells driven by the BRAF V600E mutations. These investigators used a new technique — the introduction of a library of DNA constructs, each of which encodes a different kinase — into tumor cells with the BRAF V600E mutation to screen for kinases that confer resistance to RAF inhibition. Using this screen, they confirmed a previous finding: that overexpression of RAF1 confers resistance to RAF inhibition. 8 They further showed that the overexpression of mitogen-activated protein kinase kinase kinase 8 (MAP3K8, or COT), which phosphorylates MEK in a RAF-independent manner, can also mediate resistance to RAF inhibitors…
(iii) a third basis for acquired resistance, one in which the activation of other pathways causes the tumor cell to become less dependent on ERK signaling. In these tumors, ERK activation remains sensitive to the RAF inhibitor. Specifically, they report that platelet derived growth factor receptor β (PDGFRβ), a receptor tyrosine kinase, is overexpressed in cellular models selected for RAF-inhibitor resistance in cell culture and in a subgroup of biopsy samples obtained from patients with progressing tumors. In the cell lines, PDGFRβ overexpression was associated with resistance to the anti-proliferative effects of the RAF inhibitor, despite continued inhibition of ERK signaling in the presence of the drug.
1.1.3 Immunological Techniques
Rosenberg has for decades been examining the use of the immune system to attack cancer cells and he has done a great deal of work specifically on melanoma. The second thrust of the recent advances has been along these lines and Rosenberg has also been a contributor.
The first recent report is by Schwartzentruber et al (2011, NEJM) wherein, along with Rosenberg, they have used a vaccination of a peptide which can recognize melanoma cells and then by increasing the T cells via an interleukin infusion they found that the result was improvement in survival of metastatic melanoma patients. We show the results below from the paper.

It should be noted that there is some improvement but still there is a very poor survival prognosis.
The second paper by Robert et al (2011 NEJM) uses another approach. They use a combination of a monoclonal antibody and a standard chemotherapeutic element. They state:
Ipilimumab, a fully human, IgG1 monoclonal antibody, blocks cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), a negative regulator of T cells, and thereby augments T-cell activation and proliferation.
The second agent is dacarbazine. Decarbazine is a classic alkylating agent and has been used before with very limited results.
The data on survival is shown below:
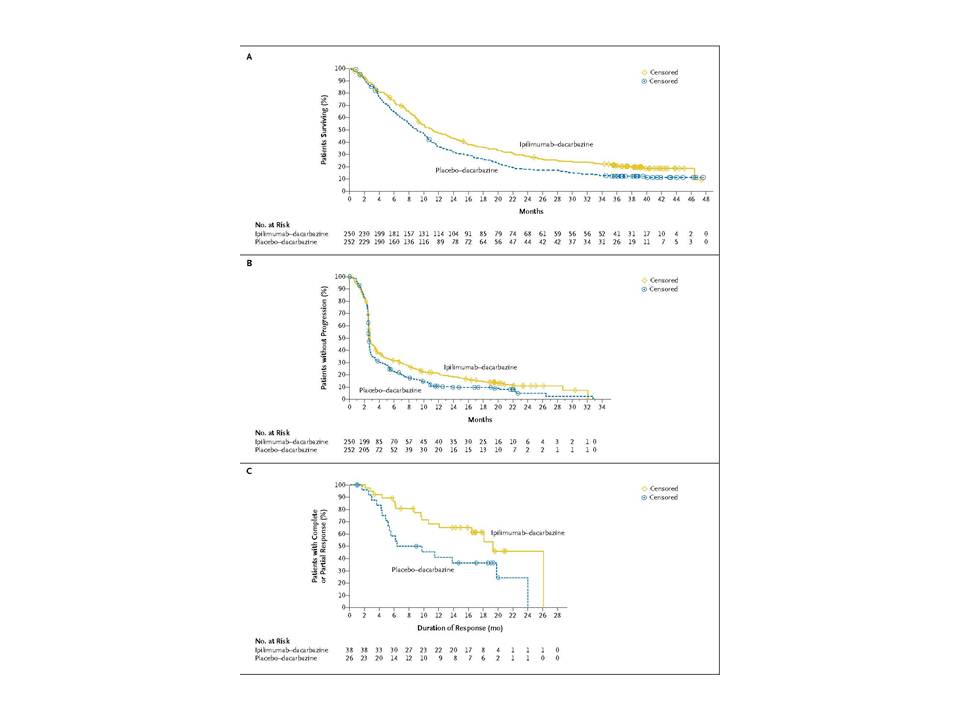
Survival with the first approach after 36 months is about 38% and with the second approach it is about 45%. The interesting factor however with the second approach is the total remission in patients exhibiting total remission at the end of the study being almost 50%. Thus if total remission was exhibited it was sustained.
As the authors of the second study state:
Prolonged survival was noted among some patients who were followed for up to 4 years. In the ipilimumab–dacarbazine group, an estimated 28.5% of the patients were alive at 2 years, and an estimated 20.8% at 3 years, as compared with an estimated 17.9% and 12.2%, respectively, in the dacarbazine group.
One can seem to state that the second approach was more effective than the first.
Possibly combining the approaches will be more effective and the current understanding is that they intend to examine those paths.
1.1.4 Considerations
The current efforts clearly show some significant advancement. However there are several key issues which must be clarified:
1. Is melanoma like colon cancer as described by Vogelstein or do we have a somewhat random set of mutations depending on the location of the lesion. Namely is melanoma really a disparate set of different sub-cancers. Is there a clear genetic pathway, is there a gene that predisposes and if so how. The how is all to often the key question.
2. Where does the melanoma stem cell fit in this paradigm? Stem cells have a problem because if they exist and are of the primary concern then perhaps we are just eliminating the TIC cells and not the CSC.
3. What of the Harahan and Weinberg model of an interacting environment? Namely what about the influence of the other parts of the body including the immune system? This has been a Rosenberg issue for decades and Harahan and Weinberg make a strong case for its consideration.
4. Is it necessary to develop a data base of aberrant expressions of proteins?
5. What about dealing with the gene itself? Why just the protein.
6. How can we identify these cells from say cell surface markers. That would enhance the ability to expand our understanding of the histology down to the expression level.
7. What genes have been changed and how? What was the change agent. We have argued elsewhere that it is radiation, ultraviolet and x-ray. But what of other factors. Where do the miRNAs fit, other epigenetic factors, methylation, and the like.
8. As with other cancers, there may be a sequence of changes, and is MIS, melanoma in situ, one of the steps. Is MIS akin to say HGPIN in prostate cancer or an adenoma in colon cancer?
There are many other issues which will evolve from this study. It represents a step in the forward direction but as has been seen each time we do this we see other new paths as unknown.
1.2 References
1. Batchelor, J., MEK and BRAF Therapy Combo Promise for Advanced Melanoma, http://www.onclive.com/conference-coverage/asco-2011/MEK-and-BRAF-Therapy-Combo-Promising-for-Advanced-Melanoma , 2011
2. Chapman, P., et al, Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation, NEJM, 2011, pp 1-9.
3. Curtin, J., et al, Distinct Sets of Genetic Alterations in Melanoma, NEJM, 2005 pp 2135-2147.
4. Eisenmann, K., et al, Mitogen Activated Protein Kinase Pathway Dependent Tumor Specific Survival Signalling in Melanoma Cells, J Can Res 2003 pp 8330-8337.
5. Ernstoff, M., Been There Not Done That – Melanoma in the Age of Molecular Therapy, NEJM, 2011 pp 1-3.
6. Flaherty, K., et al, Inhibition of Mutated Activated BRAF in Metastatic Melanoma, NEJM, 2010, pp 809-819.
7. Hanahan, D., R. Weinberg, Hallmarks of Cancer: The Next Generation, Cell, 2011, pp 646-674.
8. Hearing V., S. Leong, From Melanocytes to Melanoma, Humana (Totowa, NJ) 2006.
9. Poulikakos, P., D. Solit, Resistance to MEK Inhibitors, Science Signalling, 2011 pp 1-2.
10. Robert, C., et al, Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma, NEJM, 2011, pp 1-20.
11. Schwartzentruber, D., et al, gp100 Peptide Vaccine and Interleukin 2 in Patients with Advanced Melanoma, NEJM, 2011, pp 2119-2122.
12. Smalley, K., K. Flaherty, Integrating BRAF/MEK Inhibitors into Combination Therapy for Melanoma, Brit Jrl Cancer, 2009 pp 431-435.
13. Solit, D., et al, BRAF Mutation Predicts Sensitivity to MEK Inhibition, Nature 2006, pp 358-362.
14. Solit, D., N. Rosen, Resistance to BRAF Inhibition in Melanomas, NEJM, 2011 pp 772-774.
15. Sumitomo, H., et al, The BRAF-MAPK Signalling Pathway is essential for cancer immune evasion in human melanoma, Jrl Exp Med 2008 pp 1651-1658.


