Cancer cells originate from normal body cells in two
phases.
(i) The first phase is the irreversible injuring of
respiration. Just as there are many remote causes of plague-heat, insects,
rats-but only one common cause, the plague bacillus, there are a great many
remote causes of cancer-tar, rays, arsenic, pressure, urethane- but there is
only one common cause into which all other causes of cancer merge, the
irreversible injuring of respiration.
(ii) The irreversible injuring of respiration is
followed, as the second phase of cancer formation, by a long struggle for
existence by the injured cells to maintain their structure, in which a part of
the cells perish from lack of energy, while another part succeed in replacing
the irretrievably lost respiration energy by fermentation energy.
Because of the morphological inferiority of fermentation
energy, the highly differentiated body cells are converted by this into
undifferentiated cells that grow wildly-the cancer cells. To the thousands of
quantitative experiments on which these results are based, I should like to
add, as a further argument, the fact that there is no alternative today. If the
explanation of a vital process is its reduction to physics and chemistry, there
is today no other explanation for the origin of cancer cells, either special or
general.
From this point of view, mutation and carcinogenic agent
are not alternatives, but empty words, unless metabolically specified. Even
more harmful in the struggle against cancer can be the continual discovery of
miscellaneous cancer agents and cancer viruses, which, by obscuring the
underlying phenomena, may hinder necessary preventive measures and thereby
become responsible for cancer cases.
Namely, the purist Warburg School asserts that cancer is solely
a metabolic disorder, even further, a mitochondrial disorder. The last sentence
should be of major concern to Warburg purists. Namely, that we should not be
distracted by cancer agents or viruses. In reality we have a great deal more
evidence of the latter than of the sine qua non of Warburg.The Warburg School has many adherents and they often reject
the many current understandings of cancer initiation and progression and rely
upon a purely metabolic hypothesis. For example, in the paper by Seyfried and
Shelton the authors conclude with:
Evidence is reviewed supporting a general hypothesis that
cancer is primarily a disease of energy metabolism. All of the major hallmarks
of the disease can be linked to impaired mitochondrial function. In order to
maintain viability, tumor cells gradually transition to substrate level
phosphorylation using glucose and glutamine as energy substrates. While cancer
causing germline mutations are rare, the abundance of somatic genomic abnormalities
found in the majority of cancers can arise as a secondary consequence of
mitochondrial dysfunction. Once established, somatic genomic instability can contribute
to further mitochondrial defects and to the metabolic inflexibility of the
tumor cells.
Systemic metastasis is the predicted outcome following
protracted mitochondrial damage to cells of myeloid origin. Tumor cells of
myeloid origin would naturally embody the capacity to exit and enter tissues.
Two major conclusions emerge from the hypothesis; first that many cancers can
regress if energy intake is restricted and, second, that many cancers can be
prevented if energy intake is restricted. Consequently, energy restricted diets
combined with drugs targeting glucose and glutamine can provide a rational
strategy for the longer-term management and prevention of most cancers[1].
This conclusion is a clear statement of cancer prevention
and even management via a metabolic mechanism, namely limitation of glucose.
While there is substantial interest in the metabolic elements of cancer,
reliance upon one thread amongst many may pose substantial risks. For example
the excess of ROS, reactive oxygen species, as may be found in inflammatory
cancer initiators may be related in a metabolic manner but are not related as
directly as that os the Warburg mitochondrial process[2].
When we examine the work regarding the Warburg effect, we
note several factors as recent research has proceeded:
1. The Warburg effect, namely a rebalancing of reduced pyruvate
fed TCA generation or ATP as compared to an enhanced lactate production is most
likely an effect of the complexity in pathways in cancer cells.
2. Cancer cells manage to readjust their metabolic systems
to enhance their growth and proliferation often in low oxygen environments.
3. There appears to be a set of well-defined pathways that
play a strong role in the switch to Warburg like metabolic proliferation.
4. Cancer cells seem to proliferate almost only in a Warburg
manner so that blockage of the adjusted pathways may be a means to starve off
the cells.
5. There does not appear to be a clearly define set of
immune system determinants to target.
Overall, one of the most comprehensive and balanced papers
regarding cancer metabolism is the 2016 paper by Pavlova and Thompson. Unlike
many of the papers which blindly support the Warburg thesis, these authors
provide a well-balanced summary of the facts and where they may lead.

There are a multiplicity of "causes" of various
cancers. We depict the usual suspects below. More than likely as the various
forms of the disease are better understood, some of these factors may become just
a consequence of the disease. Some may be a cause, a consequence, and a random
coincidence.Our focus herein is the metabolic path although we have
considered many of the others in some detail. The metabolic path typically is
broad in cancers covered. Unlike say the epigenetic paths where we have a
collection of specific and identifiable cancers related to specific
alterations, the Warburg supporters throw a broad net across almost any and all
cancers. In effect the world view of those on the metabolic front often reflect
a more classic early 20th century view of cancers, namely a
commonality amongst all organs.
Energy in a cell is generated by the conversion of glucose
to ATP. ATP then is a molecule which can give up a phosphate and release
energy. That energy then is used throughout the cell. Thus ATP dynamics is at
the heart of the metabolic process. We provide but the briefest overview here
so as to allow recall. There is a significant amount of literature on these
topics[3].
Cells require a continual source of energy. One primary
source is glucose, but there are many other sources as well. As regards to
glucose as it applies to Warburg, there are three generally accepted paths in
which we see the breakdown of glucose. Fermentation and its product alcohol
will not be examined. However the water and carbon dioxide path is the oxygen
consuming path of normal cellular metabolism and the lactate path is the oxygen
depleted path. Warburg came to the conclusion of a third or if you will a
fourth (assuming you include fermentation) type of path, one which uses some
oxygen but not that much. It was the depleted aerobic path that is the heart of
the Warburg construct.
The two classic paths that we focus upon are shown below:
with and without oxygen.
 In contrast the Warburg path is a blend og two. Note that it
somehow uses both approaches.
In contrast the Warburg path is a blend og two. Note that it
somehow uses both approaches.
 The explanation for this bifurcation seems
obscure. Later we attempt to provide a rationale that can explain it. Note that both paths work but the classic oxygen path only
slightly. The ATP production is slightly better than the anaerobic path. The
details as to how this bifurcated pathway mechanism functions is yet to be
determined. One key question which should be kept in mind is: Can the genes
controlling this pathway be themselves controlled and then if this pathway is
then disturbed does it have a therapeutic effect on cancer cells. If Warburg
effects are merely consequential the answer is there is no effect. If the
answer is that there is a therapeutic effect, then Warburg may be correct.
The explanation for this bifurcation seems
obscure. Later we attempt to provide a rationale that can explain it. Note that both paths work but the classic oxygen path only
slightly. The ATP production is slightly better than the anaerobic path. The
details as to how this bifurcated pathway mechanism functions is yet to be
determined. One key question which should be kept in mind is: Can the genes
controlling this pathway be themselves controlled and then if this pathway is
then disturbed does it have a therapeutic effect on cancer cells. If Warburg
effects are merely consequential the answer is there is no effect. If the
answer is that there is a therapeutic effect, then Warburg may be correct.


We will now examine some details on each part of these.The classic path goes from glucose to water and carbon
dioxide. It is a very efficient path and energy rich pumping out well over 30
ATP molecules per glucose molecule. Thus it is the path we see in normal
metabolism. An overall summary of the key elements is shown below. This
includes three fundamental steps: (i) glycolysis which is the breakdown of
glucose to pyruvate producing 2 ATP per glucose molecule, (ii) TCA cycle
producing ATP and its precursors NAD and FAD related molecules, (iii) the
proton pump mechanism which converts NAD and FAD to ATP. Phosphorylation is a
key process whereby energy is transferred. We see this in many pathways and
this is but one.


Glycolysis is shown below where we go to pyruvate and then
to lactate. We do not present the full details since they are well known and
available elsewhere[4]. In a similar fashion we show below the simplified version of
the TCA or Krebs cycle below. We have not shown the inputs and the outputs but
only the main elements[5].
This is the most productive of all parts of this process in the direct and
indirect production of ATP and it is the section that requires oxygen. Without
oxygen this cycle does not function. The combinations above produce 32 to 36 ATP molecules. This process is the previous one but with no operative TCA.
This is because there is no oxygen available. Thus we are limited with ATP
generated solely by glycolysis.
Aerobic Glycolysis is the
Warburg construct. We have shown it above. The current understanding is that it
is a small amount of the oxygen based cycle and a dominant amount of the oxygen
poor cycle. This is a cycle that has some small oxygen contribution, that a small
contribution from a TCA and is dominated by the first step glycolysis.
What we reviewed above was the
three mechanisms of ATP generation. However we have not described their
dynamics, namely the rate at which each of these processes act. Let us look at
oxidative phophorylation, the classic path if you will, as a two step process.
Namely step one is glycolysis and step two is TCA combined with the proton
pump. Let us further assume we can get 2 ATP from glycolysis and 32 from the
second process. That is fine but what of the dynamics. How many ATP can we get
per second for example. If we have a very "hungry" cell and a rich
glucose environment, or even a cell which can scavenge its environment better
than any others, the cell will have lots of food and then it starts the
process. However we are faced with a rate problem. If step 1, glycolysis, can
just run at any rate, say a rate R, which generates say R ATP cycles per
second, or 2 times R actual ATP, then we have a fast generator. Now assume the
second path combined can run at a much lower rate. It can generate 32 for each
cycle but it is limited to a maximum number of cycles per second which is
substantially less that the glycolysis. We have the following model:

Now this is the rate analysis. It
is the maximum rate analysis. If the rate of the second steps is say three
orders of magnitude that of the first step, then the effective rate dominates
and the process pumps out ATP and the second steps does the same at a higher
number but at a much smaller rate. Thus the total number is dominated by the
first and rate limited by the second. This is similar to an enzymatic rate
limited system. In fact this totally explains the Warburg effect.

We would now have to examine the
driver of this process. Namely the supply of glucose. We assume a glucose
supply of S molecules per second. We further assume a capacity C for each step
in terms of cycles per second, namely processing a glucose molecule or its
product.
 We also assume a yield Y of so many ATP per cycle.Then:
We also assume a yield Y of so many ATP per cycle.Then:


We can depict this below:


Now let us consider a simple
example. Choose the following constants:
Note the yield is 4 ATP per glucose. This is a model which explains the Warburg effect. Namely
there are rate limiting steps in the glycolysis, TCA proton pump model and
ultimately the fastest controls. In fact if we make this fast enough we drive
it to at best 2 ATP per glucose and that is just for the glucose that are
processed.
We can now take this a step further and examine the rate
processes. We know from the Gibbs free energy results how well each reaction
works. We also have data on reaction rates which we can use for each step. Then
we can use classic reaction rate theory to ascertain if the data used in the
above example is reflective and what changes should be made.
Let us look at classic reaction rate example.

We assume we know the reaction rate for each reaction. Let
us focus just on the three step example. We can then extend it to the
glycolysis and TCA directly. We can follow Moore (pp 345-347) for this simple
example. For reactions we have:

Thus for rate equations we have given the rate constants k:

Moore then solves these equations as follows:
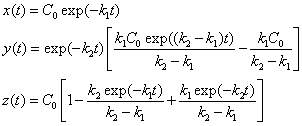
We can now perform a simple analysis with these equations to
demonstrate one more detail below in a simplified example. Here we plot the
concentrations of A, B and C as a function of time. Initially we have all A,
say glucose. Then an intermediate B is generate but it is used up, and then C
the end product appears. The time required to go from A to C is the critical
factor. We could then apply this to the glycolysis and TCA chains and
demonstrate such rate limiting. It should be noted that we can use IUPAC data
on reaction rates for each element in these chains as contained in the Serjeant
and Dempsey tables. We have done this for a few steps and it appear achievable
and can then be used for verification.Note that [A] decays exponentially, [B] increases and then
decreases, and then [C] reaches a maximum. This concept then applies for the
speed of any one of the three elements.

Otto Warburg studied the process of cancer in terms of the
ATP generation under limited or no oxygen conditions. To an extent this was the
state of fermentation, but not one leading to the production of alcohol. As for
his background, the details on the Nobel Prize site state[6]:
Warburg was born on October 8, 1883, in Freiburg, Baden.
His father, the physicist Emil Warburg, was President of the Physikalische
Reichsanstalt, Wirklicher Geheimer Oberregierungsrat. Otto studied chemistry
under the great Emil Fischer, and gained the degree, Doctor of Chemistry
(Berlin), in 1906. He then studied under von Krehl and obtained the degree,
Doctor of Medicine (Heidelberg), in 1911. He served in the Prussian Horse
Guards during World War I. In 1918 he was appointed Professor at the Kaiser
Wilhelm Institute for Biology, Berlin-Dahlem. Since 1931 he is Director of the
Kaiser Wilhelm Institute for Cell Physiology, there, a donation of the
Rockefeller Foundation to the Kaiser Wilhelm Gesellschaft, founded the previous
year.
His award of the Nobel declares:
"for his discovery of the nature and mode of action
of the respiratory enzyme" in the field of cell physiology, metabolism for
the work as follows:
In our cells nutrients are broken down so that energy is
released for the construction of cells. This respiration requires enzymes,
substances that facilitate the process without being incorporated in the final
products. Otto Warburg studied the respiration of sea urchins and other organisms
at an early stage of development. By measuring oxygen consumption in living
cells and studying which enzymes reacted, in 1928 he concluded that the
respiration enzyme he was looking for was a red ferrous pigment related to the
blood pigment, hemoglobin.
Thus despite having been awarded the Nobel, it was not for
his work in cancer. Yet Warburg seems most well-known for his cancer
conjectures. In Warburg's 1956 Science paper he begins by noting:
Since it is known how much adenosine triphosphate can be
synthesized by respiration and how much by fermentation, we can write
immediately the potential, biologically utilizable energy production of any
cells if we have measured their respiration and fermentation. With the ascites
cancer cells of the mouse, for example, we find an average respiration of 7
cubic millimeters of oxygen consumed per milligram, per hour, and fermentation
of 60 cubic millimeters of lactic acid produced per milligram, per hour. This,
converted to energy equivalents, means that the cancer cells can obtain
approximately the same amount of energy from fermentation as from respiration,
whereas the normal body cells obtain much more energy from respiration than
from fermentation. For example, the liver and kidney of an adult animal obtain
about 100 times as much energy from respiration as from fermentation.
In effect, Warburg examined cells from ascites, the fluid produced frequently from metastasized cancer in the liver. He then examined how much lactate is produced, namely the end product of anaerobic production of ATP. Strangely is appeared that these cancer cells had a combination of anaerobic plus normal metabolism which he called aerobic. Thus Warburg noted that cancer cells almost exclusively obtain their energy not from a TCA method primarily but from some small mix of a TCA plus mostly but not exclusively from what he termed fermentation, or anaerobic paths. Warburg then concluded that this aerobic path was the "cause" of cancer, not just an artifact of a cancerous process.
Warburg then goes on to state:
Clinical experiences along these lines are innumerable: the
production of cancer by intermittent irritation of the outer skin and of the mucosa
of internal organs, by the plugging of excretory ducts of glands, by cirrhosis
of tissues, and so forth. In all these cases, the intermittent irritations lead
to intermittent circulatory disturbances., Probably chronic intermittent oxygen
deficiency plays a greater role in the formation of cancer in the body than does
the chronic administration of respiratory poisons. Any respiratory injury
due-to lack of energy, however, whether it is produced by oxygen deficiency or
by respiratory poisons, must be cumulative, since it is irreversible. Frequent
small doses of respiratory poisons are therefore more dangerous than a single
large dose, where there is always the chance that the cells will be killed
rather than that they will become carcinogenic.
Frankly there is no basis for any of the above assertions.
Warburg's observations of lactate excess in cells which may have an abundance
of oxygen, and primarily cancer cells, is just that, an observation. The
generalizations emanating therefrom are at best speculation. As we shall note,
however, there has arisen an almost cult like group who have taken Warburg's
observations and correlations to extremes.
Notwithstanding the speculation, the observation clearly has
merit, merit as a distinguishing characteristic. However the statements made by
Warburg such as:
It follows from this that there would be no cancers if
there were no fermentation of normal body cells, and hence we should like to
know, naturally, from where the fermentation of the normal body cells stems and
what its significance is in the body. Since, as Burk has shown, the
fermentation remains almost zero in the regenerating liver growth, we must
conclude that the fermentation of the body cells has nothing to do with normal
growth.
On the other hand, we have found that the fermentation of
the body cells is greatest in the very earliest stages of embryonal development
and that it then decreases gradually in the course of embryonal development.
Under these conditions, it is obvious-since ontogeny is the repetition of
phylogeny-that the fermentation of body cells is the inheritance of undifferentiated
ancestors that have lived in the past at the expense of fermentation energy.
Warburg's claim of "ontogeny recapitulates
phylogeny" can be rephrased as "what comes first, the chicken or the
egg?" For one must accept the Bacon like observation of this metabolic
effect, however one must be wary of Galen like logic that is phenomenologically
limited.
We first examine some current understandings of the classic
Warburg hypothesis. As Liberti and Locasale note:
During the 1920s, Otto Warburg and colleagues made the
observation that tumors were taking up enormous amounts of glucose compared
with what was seen in the surrounding tissue. Additionally, glucose was
fermented to produce lactate even in the presence of oxygen, hence the term
‘aerobic glycolysis’. However, it was also noted that respiration alone could maintain
tumor viability. Therefore, it was concluded that, to kill tumor cells by
depriving them of energy, both glucose and oxygen had to be eliminated.
Subsequently, in 1929, an English biochemist, Herbert Crabtree, extended
Warburg's work and studied the heterogeneity of glycolysis in tumor types.
Note that the conclusion regarding killing cancer cells was
via starvation. Unfortunately that would likely cause death of all cells around.
He confirmed Warburg's findings, but further discovered
that the magnitude of respiration in tumors was variable, with many tumors
exhibiting a substantial amount of respiration. Therefore, Crabtree concluded
that not only do tumor cells exhibit aerobic glycolysis, but that there is also
variability in fermentation, presumably due to environmental or genetic
influences. Contrary to the findings of these previous works and for reasons
unclear to these authors, Warburg later proposed that dysfunctional
mitochondria are the root of aerobic glycolysis. Warburg further hypothesized
that this event is the primary cause of cancer.
The dysfunctional mitochondria is one os a possible number
of explanations. As we have discussed and shown, another explanation could be
rate limiting processes, and one suspects there are other mechanisms as well.
This phenomenon was then termed the Warburg Effect during
the early 1970s by Efraim Racker, who also pointed out that previous data
showed respiratory capability of tumors. Racker developed his own theories
about the origins of the Warburg Effect, ranging from imbalances in
intracellular pH to defects in ATPase activity. It was later observed by
Racker, Jeffrey Flier, and Morris Birnbaum that aerobic glycolysis was a controllable
process that can be directly regulated by growth factor signaling.
By that time, the discovery of oncogenes led to the
conclusion that aberrant regulation of growth factor signaling is an initiating
event in oncogenesis. Thus, their observations brought newfound significance to
Warburg's hypothesis in cancer biology. Nevertheless, it remained unclear
whether the Warburg Effect was a bystander in cancer pathogenesis until more
recently, when genetic and pharmacological studies conclusively showed that the
Warburg Effect was required for tumor growth.
Coming back to the original findings on tumor metabolism,
it is now apparent that targeting both aerobic glycolysis and mitochondrial
metabolism may be required . Throughout this history, the function of the
Warburg Effect has remained controversial. Here, we discuss several of the
major proposals and argue that the functions of the Warburg Effect for tumor
growth remain unknown even today.
Indeed it is a controversial observation. We will discuss
latter the work by some who see this as a panacea for cancer treatment. Namely
the massive blocking of glucose and in turn the aerobic metabolic pathway. As a
side note, it is also know that excess lactate has deleterious effects, but
that may at best be a sidebar.
We now examine the current view. We start with the recent
work of Levine and Kutter who note:
Rapidly dividing cells require favorable energetics, that
is, higher ATP/adenosine diphosphate (ADP) and ATP/adenosinemonophosphate (AMP)
ratios. Many cancer cells satisfy this problem by taking up much larger amounts
of glucose than do normal cells. This results from facilitated glucose transport
by one or more of several isozymes of membrane glucose transporters (GLUT 1 to
9). Once inside the cell, glucose is phosphorylated by one of several
hexokinase enzymes (the first step in glycolysis) to keep it in the cell
because of the charge added to glucose. The high concentrations of glucose in
the cells of a cancer may be observed by positron emission tomography (PET)
scans of radioactive F-19-2-deoxyglucose (FDG is not metabolized but is located
in the cell), which is indicative of enhanced glucose uptake by cells.
Many, but not all, cancers have this property of
increasing glucose uptake, and this is a confirmation of the Warburg effect.
With large amounts of glucose available in a cell, glucose is metabolized
through the PPP, producing nucleosides and generating NADPH. The NADPH is
essential for fatty acid synthesis, along with acetyl-CoA (which is made from
some of the pyruvate in mitochondria that is not converted to lactate). NADPH
also contributes to a proper redox control and protects the cell from ROS.
There are several ways the cell responds to lower ROS (reactive oxygen species)
levels, but by far the major molecule involved is glutathione (GSH), which
eliminates ROS by accepting an electron and is converted to its oxidized form,
GSSG (glutathione disulfide).
The enzyme glutathione reductase uses NADPH to reduce
GSSG to GSH. Thus, NADPH is a major source of cellular “coolant” when oxidative
reactions run too “hot” (high ROS levels) by using large amounts of glucose to
produce both substrates and energy. However, high levels of ROS can be
advantageous for cancer cells when they allow for the stimulation of cell
proliferation, induction of genetic instability, and evasion from senescence.
As Thompson, who has done extensive recent work in cancer
cell metabolism, noted (see Riccio):
After the initial step in glucose metabolism—glycolysis,
conversion of glucose to two molecules of pyruvate—mitochondrial oxidative
phosphorylation usually proceeds to yield ATP. But even in the presence of
oxygen, many cancer cells divert pyruvate to fermentation, producing lactate.
This less rewarding mode of ATP production demands a relatively high rate of
glycolysis. Otto Warburg described this shift toward "aerobic
glycolysis" in cancer cells in 1924. The molecular and genetic basis of
the Warburg effect, however, has only recently come to light. Contrary to
Warburg's hypothesis that mitochondrial defects necessitate this shift, most
cancer cells maintain the ability to execute oxidative phosphorylation and do
fully catabolize a small amount of glucose.
Cancer cells are genetically differentiated from normal
cells, but it is now clear that the metabolic shifts they exhibit are also
partly required for division of normal cells. In a quiescent cell, maximum ATP
production yields enough energy for cellular machinery, and at least 50% of
free energy is used for ion transport across the membrane. When a cell divides,
glycolytic intermediates are diverted from the tricarboxylic acid (TCA/Krebs)
cycle to reserve carbon and nitrogen for fatty acid synthesis and for
production of nonessential amino acids. DNA replication demands de novo
nucleotide synthesis, beyond the supply garnered from recycling pathways in a
non-dividing cell. Ribose, serine, and glycine (byproducts of glucose
metabolism), as well as glutamine for pyrimidine production, are needed for nucleotide
synthesis.
Finally Thompson notes the active role genetic alterations
play. This is sharp contrast to the abject rejection of such by Warburg and his
followers. Specifically Thompson notes:
The most commonly mutated gene in cancers is KRAS. The KRAS
protein, a GTPase, normally functions as a molecular switch, relaying signals
received by receptor tyrosine kinases and other receptors of extracellular
signals. Two of its main targets include the MAPK and PI3K signal transduction
cascades. But many indirect targets of KRAS are involved in cellular
metabolism, including glucose transporters that are positively regulated by the
PI3K/Akt pathway. Glutamine-addicted tumors are often characterized by the
oncogenic expression of Myc, a transcription factor that promotes the
expression of glutamine transporters as well as metabolic enzymes needed for
biosynthesis. Constitutively activated KRAS thus primes a cell to undergo
aerobic glycolysis by ensuring a steady influx of glucose.
Namely the Warburg effect is not standard for cancer cells.
Thus the mass of conclusions based upon this faulty construct a truly built
with feet of sand. We shall detail this further.
Warburg factors relate to the metabolism of the cell,
focusing on the mitochondria. However there are many cell factors which in turn
control this process for better or worse. There has been a great deal of study
examining the various genetic pathways and their controls on the metabolic
actions that are a focal point of the Warburg effect. The following Figure
depicts the complex interaction of pathways and metabolism.
The paper by DeBerardinis et al from which we have
abstracted the above demonstrates several of the pathways control elements which
may be prominent in controlling the metabolic processes. They state regarding
the above diagram:
The model shows some of the prominent aspects of
metabolism in proliferating cells, including glycolysis; lactate production;
the use of TCA cycle intermediates as macromolecular precursors; and the
biosynthesis of proteins, nucleotides, and lipids. The PI3K/Akt/mTOR pathway,
HIF-1a, and Myc participate in various facets of this metabolic phenotype. The
binding of a growth factor (GF) to its surface receptor brings about activation
of PI3K and the serine/threonine kinases Akt and mTOR.
Constitutive activation of the pathway can occur in
tumors due to mutation of the tumor suppressors PTEN, TSC1, and TSC2, or by
other mechanisms. Metabolic effects of the PI3K/Akt/mTOR pathway include
enhanced uptake of glucose and essential amino acids and protein translation.
The transcription factor HIF-1a is involved in determining the manner in which
cells utilize glucose carbon. Translation of HIF-1a is enhanced during growth-factor
stimulation of the PI3K/Akt/mTOR pathway. In the presence of oxygen, HIF-1a is modified by prolyl
hydroxylases, which target it to a ubiquitin ligase complex that includes the
tumor suppressor VHL. This association results in constitutive normoxic
degradation of the HIF-1a protein. Hypoxia, mutation of VHL, or accumulation of
reactive oxygen species (ROS) or the TCA cycle intermediates succinate and
fumarate impair HIF-1a degradation, allowing it to enter the nucleus and engage
in transcriptional activity. Transcriptional targets include genes encoding
glucose transporter 1 (GLUT1), LDH-A, and PDK1. The combined effect on glucose
metabolism is to increase both glucose utilization and lactate production, as
PDK1 inhibits conversion of pyruvate to acetyl-CoA by pyruvate dehydrogenase
(PDH). The transcription factor Myc increases expression of many metabolic
enzymes, including glycolytic enzymes, LDH-A, and several enzymes required for
nucleotide biosynthesis.
Abbreviations: PI3K, phosphatidylinositol 3-kinase; PTEN,
phosphatase and tensin homolog; TSC, tuberous sclerosis complex; mTOR,
mammalian target of rapamycin; glc-6-P, glucose-6-phosphate; 3-PG,
3-phosphoglycerate; PDK1, pyruvate dehydrogenase kinase 1; SDH, succinate
dehydrogenase; FH, fumarate hydratase; HIF-1a, hypoxia-inducible factor 1a;
VHL, von Hippel-Lindau.
Now there are multiple other papers which depict variants on
the above and referenced herein. One suspects that this analysis is still a
work in progress and as such we expect a considerable amount of complexity. The
full details of the interaction and control are yet to be specified. This does
raise the issue; is the Warburg effect an artifact or a separate entity? The
observations of it being an artifact are currently compelling.
Thompson in EMBO notes regarding the work of Pate et al:
In this issue of The EMBO Journal, Pate et al identify
Wnt signaling as a mechanism that suppresses pyruvate oxidation in the TCA
cycle and promotes rather than inhibits cell proliferation. As such, Wnt
signaling is a candidate for the signal transduction pathway that could
synergize with PI3K/AKT signaling in proliferating cells. Wnt signaling is
already well characterized as a regulator of cell proliferation.
First, Wnt induced LEF/TCF/b-catenin transcription
complexes have been implicated in controlling cell proliferation through the
induction of Myc and Cyclin D. Cyclin D levels are critical to cell cycle
progression through G1, and Myc has been implicated in the stimulation of
glutamine metabolism and nucleotide synthesis necessary to support S-phase.
Pate et al identify additional transcriptional targets of LEF/TCF/b-catenin
complexes.
They report that the genes connected to metabolism are
the most highly overrepresented category of Wnt-target genes. Two relevant Wnt
targets were identified: pyruvate dehydrogenase kinase 1 (PDK1) and the lactate
transporter (MCT-1). These two proteins along with the Myc induced gene LDH-A
allow Wnt-activated cells to divert glycolytic pyruvate away from the TCA cycle
by converting it into lactate and promoting lactate secretion from the cell.
Induction of PDK1 was found to be required for Wnt-induced aerobic glycolysis,
in vivo tumor cell accumulation, and VEGF independent angiogenesis.
Pate et al did not
test for whether Wnt induction of Myc and/or glutamine metabolism contributes
to Wnt-induced tumor cell accumulation. However, it is reasonable to suspect
that Wnt induction of PDK1 cooperates with Myc-induced glutaminolysis to
facilitate a cellular transition from growth to proliferation since Myc is a
well-characterized effector of Wnt signaling.
The combined effects of Wnt-facilitated aerobic
glycolysis and Myc-induced glutaminolysis provide the cell with a potent
ability to engage in de novo nucleotide biosynthesis
This observation would modify the above diagram by adding a
WNT receptor and having it drive MYC. Thus we now would understand the driver
of MYC and the importance of the WNT connection. This is an added example of
the progression of the modifications and understanding of the metabolism which
seems to be tightly controlled by various pathways, and pathways which we know
to be subject to attack in many cancers. Thus the argument that the Warburg
proponents make that the pathways are irrelevant seems to be destroyed in these
studies.
Finally in a recent paper by Pavlova and Thompson we have:
Tumorigenesis is dependent on the reprogramming of
cellular metabolism as both direct and indirect consequence of oncogenic
mutations. A common feature of cancer cell metabolism is the ability to acquire
necessary nutrients from a frequently nutrient-poor environment and utilize
these nutrients to both maintain viability and build new biomass. The
alterations in intracellular and extracellular metabolites that can accompany
cancer-associated metabolic reprogramming have profound effects on gene
expression, cellular differentiation and the tumor microenvironment.
In this Review, we have organized known cancer-associated
metabolic changes into six hallmarks:
(1) deregulated uptake of glucose and amino acids,
(2) use of opportunistic modes of nutrient acquisition,
(3) use of glycolysis/TCA cycle intermediates for
biosynthesis and NADPH production,
(4) increased demand for nitrogen,
(5) alterations in metabolite-driven gene regulation, and
(6) metabolic interactions with the microenvironment.
While few tumors display all six hallmarks, most display
several. The specific hallmarks exhibited by an individual tumor may ultimately
contribute to better tumor classification and aid in directing treatment.
In the above paper the authors fill in further details of
internal gene pathway controls on the cell metabolic process.
We can now make a few observations which have merit when examining
the Warburg effect.
1.
If cancer cells need glucose to produce ATP, but they
produce at best 4 ATP per glucose, whereas normal cells produce 30-36, then if
we reduce glucose dramatically, to a basal amount for normal cells, would that
"starve" cancer cells. Example is metformin and prostate cancer.
This has been the standard conjecture that advocates of
Warburg effects articulate. As Levine and Kutter noted:
Cells from some tumors use an altered metabolic pattern
compared with that of normal differentiated adult cells in the body. Tumor
cells take up much more glucose and mainly process it through aerobic
glycolysis, producing large quantities of secreted lactate with a lower use of
oxidative phosphorylation that would generate more adenosine triphosphate
(ATP), water, and carbon dioxide.
This is the Warburg effect, which provides substrates for
cell growth and division and free energy (ATP) from enhanced glucose use. This
metabolic switch places the emphasis on producing intermediates for cell growth
and division, and it is regulated by both oncogenes and tumor suppressor genes
in a number of key cancer-producing pathways. Blocking these metabolic pathways
or restoring these altered pathways could lead to a new approach in cancer
treatments.
For example, we have examined the use of metformin in
prostate cancer and the result can be a diminution of the malignancy.
2.
Since we have internal cell pathways to control metabolic pathways
can we identify specific proteins to block to starve the aerobic pathway? If
so, then what harm may that cause other than starving the cancer?
Pathway control of the Warburg effect has been examined by
several recent studies. We have referred to them and have discussed them in
some detail. However their use is still and open question. Secondary harms are
all too often the controlling factor. By starving the patient of glucose do we
create a plethora of secondary and unintended but deleterious consequences? One
suspects that to be the case.
As Tisdale noted when examining cachexia in cancer:
Most cancer cells use glycolysis as the principal method
to generate ATP, and this phenomenon is called the Warburg effect. The
increased glucose uptake by tumors is the basis of the [18F]fluorodeoxyglucose
positron emission tomography (FDG-PET) tumor diagnostic method, which is based
on the assumption that cancer tissue has a higher rate of glucose uptake than
normal tissue (29). In addition, glycolytic inhibitors have been suggested as
being useful to specifically target the slow-growing cells of a tumor, which
would complement currently used chemotherapeutic agents and radiation, which
target rapidly growing cells. Several reasons have been suggested to explain
this phenomenon including dysfunctional mitochondria, which exhibit frequent
mutations in the DNA which would prevent their use of the tricarboxylic acid
cycle, preventing the total combustion of pyruvic acid. Since mitochondrial DNA
codes for 13 components of the respiratory chain, it is likely that such
mutations would cause malfunctions in respiration. Indeed,
respiration-deficient cells with deletions in mitochondrial DNA show an
increased dependency on glycolysis, increased NADPH and activation of the Akt
survival pathway, resistance to antitumor drugs, and a survival advantage in
hypoxic conditions. Other alterations include overexpression of the “low Km”
form of hexokinase, type II hexokinase, due to gene demethylation, resulting in
tumor glucose utilization at normal blood sugar levels, oncogenic signals, such
as ras and src, which increase dependence on glucose, and tumor hypoxia due to
growth beyond the vascular supply. Hypoxia activates a transcription factor
called hypoxia- inducible factor 1 (HIF-1), which increases the transcription
of the cell-surface glucose transporter GLUT1, and at least one isoform of
nearly all the core enzymes of glycolysis.
Overall Tisdale does not relate Warburg that strongly with
the overall cachexia process. To some degree this is surprising.
3.
Is there a Reverse Warburg Effect?
Can Warburg work in reverse? As Xu et al note:
There is much evidence that the Warburg effect has many
questionable points. Based on a mass of research, a new hypothesis is catching
people’s attention, the reverse Warburg effect. Glycolysis occurs in
mesenchymal stoma cells under the activation of neighboring cancer cells.
Furthermore, an increased formation of recycled nutrients is produced. This
high-energy metabolism is transferred to the neighboring cancer cells by the
orientation of transport to participate in the TCA cycle.
The consequence is that the OXPHOS (oxidative
phosphorylation) increases enhancing ATP production, thus constituting
metabolic coupling. This new model may well explain both the way ATP is
produced via a low efficiency method despite extremely high energy demand of
the tumor cells, and reasonably explain the ‘autophagy paradox’ that has long
been questioned. Although our focal point on the aerobic glycolysis mirrors the
core of the realm, further research is still required on cancer bioenergetics.
We have argued that there is a simpler way to explain
Warburg, namely simple rate limiting.
4.
Epigenetics can be induced by metabolic pathways. How then does the
metabolic issue in and around cancers play with the epigenetic changes?
Epigenetics has become one of the fertile areas for
understanding cancers. We use herein a broad definition of epigenetics as one
where there is a non-DNA specific factor altering the ultimate expression of a
gene. This may range from simple DNA methylation, methylation of acetylation of
chromosome complexes, micro RNAs and other related gene control mechanisms. As
Yun et al note:
Epigenetics is
defined as heritable changes in
gene expression without alterations in
the underlying genetic material. Modifications
include DNA methylation and covalent post-translational modifications of
histones such as acetylation,
methylation, phosphorylation, ubiquitination, phosphorylation, and
crotonylation. Since every cell in the body has the same genetic code, epigenetic regulation of gene expression
plays a large role in determining cellular identity. Failure of proper maintenance of cellular
epigenetic status can, thus, result in loss of tissue identity or aberrant signaling pathways that
lead to developmental defects or dis- ease states such as diabetes and cancer .
It is now well accepted that cancer initiation and progression are driven by a
series of genetic and epigenetic alterations that cause either activation of oncogenes or
inactivation of tumor suppressor genes.
Much of the recent excitement in the
field of cancer epigenetics lies in the reversible nature of epigenetic alterations; unlike genomic mutations, these
changes can theoretically be reversed by epigenetic therapy.
Recently, four drugs that target the epigenetic machinery
have been approved by the FDA for cancer
treatment and have demonstrated prolonged survival and lower toxicity than conventional chemotherapy . Despite intensive
research and remarkable advances in our
understanding of epigenetics, the mechanisms
and regulators that trigger
pathological epigenetic reprogramming in
cancer remains poorly understood.
How these relate to Warburg is unclear. However Bensinger
and Christofk have noted regarding miRNA and Warburg the following:
Since the discovery that miRNAs are aberrantly expressed
in cancer, accumulating evidence suggests that miRNAs contribute to tumor
growth by modulating levels of oncogenes and tumor suppressors. Not
surprisingly, some miRNAs have been shown to regulate cancer metabolism.
miRNA-23a and miRNA-23b, which are suppressed by MYC, repress mitochondrial
glutaminase expression. Therefore, MYC enhances glutaminase and glutamine metabolism,
an important carbon and nitrogen source for biosynthesis in cancer cells, by
repressing miRNA-23a/b expression. A recent study by Eichner et al. found that
ERBB2 signaling leads to miRNA-378 expression, which promotes the Warburg
effect by inhibiting expression of ERR, a binding partner for PGC-1, leading to
reduced transcription of tricarboxylic acid cycle genes. miRNA- 378 expression,
which correlates with progression in human breast cancer tissues, causes
increased lactate production, decreased respiration, and increased
proliferation of breast cancer cell lines. Another recent study has implicated
miRNA-210 in metabolic reprogramming in cancer….miRNA-201, which is induced by
hypoxia, represses the mitochondrial iron sulfur scaffold protein ISCU
resulting in decreased mitochondrial complex 1 activity, aconitase activity,
increased lactate production and hypoxic cell survival. Future studies will
undoubtedly uncover additional miRNAs important for aerobic glycolysis in cancer.
Thus perhaps epigenetics via the mi RNA path may have a
significant role to play.
Pavlova and Thompson also examine the interaction with the epigenetic
elements. Namely they state:
Aberrantly activated growth and survival signals that
drive tumorigenesis facilitate the reprogramming of cancer cell metabolism to
enable increased nutrient acquisition and biosynthesis. However, metabolic
networks themselves are not merely passive recipients of growth signals, but
quite the contrary, directly transmit the information about the cellular
metabolic state to a diverse array of regulatory enzymes, among which are those
that mediate the deposition and removal of epigenetic marks from chromatin. A
key metabolite that builds up when cells metabolize more glucose than needed
for bioenergetic support is cytosolic acetyl-CoA. Cytosolic acetyl-CoA is the
obligate substrate for enzymes that acetylate histones and other proteins. The
deposition of acetyl marks on histones is associated with the increased
accessibility of the genomic DNA for the assembly of transcriptional complexes,
and has a rapid turnover rate. Histone acetylation is exquisitely sensitive to
alterations in the cellular nutritional and signaling status. Indeed,
withdrawal and re-addition of glucose, as well as activation of oncogenic
signaling via introduction of an oncogenic KRAS mutant or a constitutively active
form of Akt, increase total histone acetylation, which, in turn, promotes the
enhanced and broader gene expression.
Overall there appears to be a rich field of examining
metabolism and epigenetics.
5.
Glutamine has a similar effect as glucose. Are the cellular dynamics
the same or similar?
Cells use both glucose and glutamine. As Altman et al note:
The maintenance of high levels of glutamine in the blood
provides a ready source of carbon and nitrogen to support biosynthesis,
energetics and cellular homeostasis that cancer cells may exploit to drive
tumour growth. Glutamine is transported into cells through one of many
transporters, such as the heavily studied SLC1A5, and can then be used for
biosynthesis or exported back out of the cell by antiporters in exchange for
other amino acids such as leucine, through the L‑type amino acid transporter 1
(LAT1, a heterodimer of SLC7A5 and SLC3A2) antiporter. Glutamine derived
glutamate can also be exchanged through the xCT antiporter for cystine, which
is quickly reduced to cysteine inside the cell. …
The expression of enzymes involved in glutamine
metabolism varies widely in cancers and is affected by tissue of origin and
oncogenotypes, which rewire glutamine metabolism for energy generation and
stress suppression. Of the two glutaminase enzymes28, GLS is more broadly expressed
in normal tissue and is thought to have a crucial role in many cancers, whereas
GLS2 expression is restricted primarily to the liver, brain, pituitary gland
and pancreas36. Alternative splicing adds further complexity, as GLS pre-mRNA
is spliced into either glutaminase C (GAC) or kidney-type glutaminase (KGA)
isoforms37–39. The two GLS isoforms and GLS2 also differ in their regulation
and activity. GLS but not GLS2 is inhibited by its product glutamate, whereas GLS2
but not GLS is activated by its product ammonia in vitro28,29. Although both
GLS and GLS2 are activated by inorganic phosphate, GLS (and particularly GAC) shows
a much larger increase in catalysis in the presence of inorganic phosphate37.
Sirtuin 5 (SIRT5), which can be overexpressed in lung cancer40, can
desuccinylate GLS to suppress its enzymatic activity41, whereas SIRT3 can
deacetylate GLS2 to promote its increased activity with caloric restriction.
The authors continue:
Ninety years ago, Warburg discovered that many animal and
human tumours displayed high avidity for glucose, which was largely converted
to lactate through aerobic glycolysis. Warburg also suggested that cancers are
caused by altered metabolism and loss of mitochondrial function. These dogmatic
views have been replaced and refined over the past several decades with the
emergence of oncogenic alterations of metabolism, appreciation of the
importance of mitochondrial oxidation in cancer physiology and the rediscovery
of the role of glutamine in tumour cell growth in addition to the pivotal role
of glucose.
In this Review, we provide an updated overview of
glutamine metabolism in cancers and discuss the complexity of metabolic
rewiring as a function of the tumour oncogenotype as well as the
microenvironment, which adds to the heterogeneity found in vivo. In certain
types of cancer, such as those driven by MYC, tumour cells seem to depend on
glutamine, and hence targeting glutamine metabolism pharmacologically may prove
beneficial. Conversely, different oncogenic drivers may result in tumour cells
that could bypass the need for glutamine.
However, targeted inhibition of some oncogenic drivers
has been reported to rewire cells to become dependent on glutamine, and hence
targeted inhibitors could be synthetically lethal with inhibition of glutamine
metabolism. Overall, the field of cancer metabolism has made considerable
progress in understanding alternative fuel sources for cancers, including
glutamine, which under specific circumstances can be exploited for therapeutic
purposes.
6.
Is there a relationship between the immune system and the Warburg
effect, and if so can it be used to address various types of cancers?
There has been a significant amount of recent work examining
the relationship between cell metabolism and the immune system. The metabolic
factors can result in complex immune response, separate from the more simple
issues of cancer cell markers.
Herbel et al have noted:
Conversion of normal cells to cancer is accompanied with
changes in their metabolism. During this conversion, cell metabolism undergoes
a shift from oxidative phosphorylation to aerobic glycolysis, also known as Warburg
effect, which is a hallmark for cancer cell metabolism. In cancer cells,
glycolysis functions in parallel with the TCA cycle and other metabolic
pathways to enhance biosynthetic processes and thus support proliferation and
growth. Similar metabolic features are observed in T cells during activation
but, in contrast to cancer, metabolic transitions in T cells are part of a
physiological process. Currently, there is intense interest in understanding
the cause and effect relationship between metabolic reprogramming and T cell
differentiation.
After the recent success of cancer immunotherapy, the
crosstalk between immune system and cancer has come to the forefront of
clinical and basic research. One of the key goals is to delineate how metabolic
alterations of cancer influence metabolism-regulated function and
differentiation of tumor resident T cells and how such effects might be altered
by immunotherapy. Here, we review the unique metabolic features of cancer, the
implications of cancer metabolism on T cell metabolic reprogramming during
antigen encounters, and the translational prospective of harnessing metabolism
in cancer and T cells for cancer therapy. …
T cells and cancer cells inexorably share metabolic
programs and preferences, and thus there is high competition for nutrients
between cancer and T cells within the tumor microenvironment. Nutrient
deprivation, increased metabolic waste, and the ability of tumors to express
inhibitory ligands impair the metabolic fitness and capacity of T cells to uptake
and utilize nutrients. Additionally, metabolism determinants of the tumor
microenvironment drive T cells to exhaustion and Treg differentiation programs
rather than Teff and Tm phenotypes leading to impaired antitumor responses.
The changes and adaptations in the tumor microenvironment
most likely are not limited to solid tumors because leukemia and lymphoma cells
have similar metabolic characteristics with solid tumors and often express
immunomodulatory ligands. In addition, lymphomas may also contain infiltrating
T cells with an exhausted phenotype similar to that identified in chronic viral
infections or solid tumors. Thus, drugs that directly target key metabolic
enzymes or their upstream regulators will likely interfere with metabolism of
both cancer and T cells in which core cell signaling and metabolic pathways
converge.
Understanding the similarities and differences of
metabolic vulnerabilities of T cells and cancer may lead to the development of
single-target or combination-based therapies to modify metabolism of the tumor
niche thereby targeting both cancer cells and immune cells. Identification of
such specific changes in oncometabolites and immuno-metabolites may define not
only novel therapeutic targets but also biomarkers for assessment of
therapeutic responses to tumor immunotherapy combined with metabolism-targeting
drugs. The ultimate goal is to design metabolism-based treatment strategies to
attack and eradicate cancer while promoting effective and sustainable
anti-tumor T cell responses.
As Elliott and Head have noted:
It is at this time we must briefly mention the work and
contributions of Paul Ehrlich and Otto Warburg. Paul Ehrlich’s magic bullet
theory has inspired many generations of scientist to explore numerous molecular
cancer therapeutics. He connected chemistry to biology and medicine; and
predicted the existence of specific cell recaptors .
Otto Warburg in the 1930s, described a link between
defects in mitochondrial physiology and tumorigenesis. He observed a
significant increase in glycolysis and lactate production in the presence of
oxygen without an increase and an occasional decrease in oxidative
phosphorylation. This phenomenon is known as aerobic glycolysis or the “Warburg
effect” and is well documented in tumor cells. The work of the above two
scietists has contributed much to the field of tumorigenesis, and those of us
in the field should be extremely grateful for their contributions.
In 2005 Gottlieb and Tomlinson did a tremendous job
reporting on mitochondrial tumor suppressors with a genetic and biochemical
update. They mention the work of Warburg, but it was 60 years after Warburg
that the first genetic evidence that might explain the mechanisms of aerobic
glycolysis was reported. There were many tumors shown to contain somatic
mutations in mitochondrial DNA (MTDNA).
It is thought that most are homoplastic and the outcome
is non-functional oxidative phosphorylation, causing cells to increase
glycolysis, the only other avenue for ATP (adenosine triphosphate) synthesis.
However, there is limited evidence that indicates mitochondrial mutations might
directly promote tumorigenesis. There are some mitochondrial proteins encoded
by nuclear genes that can be tumor suppressors, some are involved in benign and
malignant tumors. Two of the proteins are the enzymes succinate dehydrogenase
(SDH) and fumarate hydratase also known as fumarase. Both of these enzymes are
involved in the Kreb’s cycle that connects glucose metabolism in the cytosol to
mitochondrial oxidative phosphorylation.
The inhibition of SDH has been linked to the induction of
the hypoxicinducible factor (HIF). HIF is a transcription factor induced under
low oxygen conditions. SDH inhibition causes an accumulation of succinate,
which transmits an oncogenic signal from the mitochondria to the cytosol, which
inhibits HIF-α prolyl hydroxylase (PHD) activity leading to the stabilization
of the HIF-1α subunit at normal oxygen levels. The result is the transcription
of genes involved in tumorigenesis, such as, the angiogenesis factor vascular
endothelial growth factor (VEGF). Therefore, succinate has been identified as a
new intracellular messenger through discovery of the mitochondrion cyto-sol
pathway. Gottlieb and Tomlinson have done a great job of discussing the link of
mitochondrial dysfunction to cancer and we will now present some important
aspects of their findings.
The TCA cycle (tricarboxylic acid cycle also known as the
Krebs cycle) is fundamental to the bioenergetics of cells, however, it is not
exactly known how TCA dysfunction leads to cancer. To address that problem,
they proposed several models. They included decreased programmed cell death
(apoptosis), increased production of reactive oxygen species (ROS), and
activetion of a hypoxia-like pathway under normoxic conditions (pseudohypoxia).
Though impossible to distinguish between these options as they interact with
each other, which leads to a complex grid of tumor regulatory systems. They
still provided evidence to support the role for each of these three models in
mitochondrial dysfunction induced tumorigenesis.
Overall the potential exists for utilizing the Warburg like responses
in conjunction with the immune system although the path is not clear at this
point. However Peng et al have recently noted:
Aerobic glycolysis (the Warburg effect) is a metabolic
hallmark of activated T cells and has been implicated in augmenting effector T
cell responses, including expression of the proinflammatory cytokine
interferon-g (IFN-g), via 3′ untranslated region (3′UTR)–mediated mechanisms.
Here, we show that lactate dehydrogenase A (LDHA) is induced in activated T
cells to support aerobic glycolysis but promotes IFN-g expression independently
of its 3′UTR. Instead, LDHA maintains high concentrations of acetyl–coenzyme A
to enhance histone acetylation and transcription of Ifng. Ablation of LDHA in T
cells protects mice from immunopathology triggered by excessive IFN-g expression
or deficiency of regulatory T cells. These findings reveal an epigenetic mechanism
by which aerobic glycolysis promotes effector T cell differentiation and suggest
that LDHA may be targeted therapeutically in autoinflammatory diseases.
7.
Thus the final question is: Why does the Warburg effect even occur?
As DeBerardinis et al note:
So why does the Warburg effect occur? Clearly, the high
glycolytic rate provides several advantages for proliferating cells.
First, it allows cells to use the most abundant extracellular
nutrient, glucose, to produce abundant ATP. Although the yield of ATP per
glucose consumed is low, if the glycolytic flux is high enough, the percentage
of cellular ATP produced from glycolysis can exceed that produced from
oxidative phosphorylation. This may be due to the high rate of ATP production
during glycolysis compared to oxidative phosphorylation.
Second, glucose degradation provides cells with
intermediates needed for biosynthetic pathways, including ribose sugars for
nucleotides; glycerol and citrate for lipids; nonessential amino acids; and,
through the oxidative pentose phosphate pathway, NADPH. So the Warburg effect
benefits both bioenergetics and biosynthesis.
What remains controversial about the Warburg effect is
why the rate of lactate production is so high when more of the pyruvate could
presumably be oxidized to enhance ATP production. One explanation is simply
that glycolysis outpaces the maximal velocity of pyruvate oxidation, so that
cells must instead eliminate pyruvate using high-flux mechanisms.
We have made several observations which can lead to a few
reasonable conclusions. They are:
1. Warburg effect is most likely an artifact of the other
elements which make up for a malignancy. The classic understanding was that the
effect was the cause of cancers even though the cause of the effect itself was
unknown.
2. Warburg effect is not a cause nor a sine qua non for
cancer and addressing its control would most likely be addressing a secondary
consequence not a causal element.
3. The Warburg effect can be explained by a rate limiting
process in the overall manner in which glucose produces ATP. In fact it may be
that the cancer cells are very "hungry" cells in need of massive
amounts of glucose that they saturate the TCA and thus perform aerobic
glycolysis.
Finally we pose the questions from Bensinger and Christofk:
1. Can the Warburg effect and cancer metabolism be
programmed? While the Warburg effect metabolic phenotype was initially identified
in cancer tissue, it is now well appreciated that rapidly dividing normal
tissues, such as ES cells and lymphocytes, employ aerobic glycolysis to meet
their energetic and biosynthetic requirements during expansion. These
observations support the notion that aerobic glycolysis is a preferred
metabolic program under conditions of rapid cellular expansion. However, it
remains unclear how the Warburg effect is initiated and maintained; these need
not be the same signals in cancer versus normal tissues which lack dysregulated
signaling. One critical signaling axis for metabolic programming of normal
cells is the PI3K/AKT/mTOR pathway downstream of growth receptors. Genetic and
pharmacologic models have clearly identified mTOR signaling in controlling
cellular growth and metabolism.
This is a clear statement of what is desired but also a
clear articulation of what we really do not know about Warburg. mTOR is a
powerful control mechanism, yet is it a driver of or driven by Warburg?
2. What is the relationship between the Warburg effect
and cancer microenvironment? ….Thus,
catabolic programs in normal cells within the tumor
parenchyma can play a pivotal role in supporting the anabolic program of
cancer. Indeed, recent studies indicate that despite robust anabolic programs
in tumors, addition of lipolytic capabilities robustly increase tumorigenesis.
Although speculative, the symbiotic relationship between normal cells and tumor
cells could help to explain cancer associated cachexia by driving a generalized
catabolic program in tumor bearing individuals.
The microenvironment is becoming significant factor on a
cancer development and proliferation. Cancer cells have a high demand for
nutrients and oftentimes those nutrients are obtained from its microenvironment
even to the extant that it cannibalizes its benign neighbors, using their
remnants as energy to grow.
3. Does aerobic glycolysis contribute to chemotherapeutic
resistance or susceptibility? Given that many cancers exhibit altered
metabolism, it should not be surprising that there has been increased effort to
therapeutically target these pathways as a means to decrease tumor growth or
alter behavior.
There is a great deal of evidence that Warburg effects do
inhibit certain chemotherapies. This is a compelling path to explore since it
may enlighten us on Warburg as well as expanding understanding of
chemotherapeutic mechanisms.
4. What is the metabolism of cancer stem cells? While the
concept of bone fide cancer stem cells (CSCs) remains controversial, there is
strong evidence to indicate that a subset of cancer cells are endowed with the
capacity to initiate tumor formation. Oft times defined as tumor-initiating
cells (TICs), these cells appear to be critical for the ability of tumors to
resist conventional radio- or chemotherapeutics and repopulate the tumor during
and after treatments. Whether TICs have distinct metabolic programs from the
bulk of tumor cells is not well established.
This last question is compelling since it indicates that
Warburg is but one presentation of cancer cell metabolism. It also presents the
argument that understanding cancer cell metabolism in the whole may provide a
fruitful approach to the ultimate control of cancers. However, and this is
critical, we must always be fearful of falling into what I have termed the
"Warburg Trap", namely the positing of the "silver bullet"
cause of all cancers. Clearly if we have discovered anything, it is that
cancers have a multiplicity of causes and ongoing support mechanisms, even in
the same organ, and even in the same cell types in that organ.
1. Aguilera et al, Vitamin C uncouples the Warburg metabolic switch
in KRAS mutant colon cancer, Oncotarget, Vol. 7, No. 30, 2016
2. Altman et al, From Krebs to clinic: glutamine metabolism to
cancer therapy, Nature Reviews, Cancer, Volume 16, October 2016, 619
3. Atkins, P. Physical Chemistry, 4th Edition, Freeman
(New York) 1990.
4. Bensinger and Christofk, New aspects of the Warburg effect in
cancer cell biology, Seminars in Cell & Developmental Biology 23 (2012)
352– 361
5. Berg, et al, Biochemistry, 5th Edition, Freeman (New
York) 1995
6. Cairns and Mak, The current state of cancer metabolism, Nature
Reviews, Cancer Volume 16, October 2016, 613
7. Cairns et al, Regulation of Cancer Cell Mechanism, Nature
Reviews Cancer, Feb 2011.
8. Currie et al, Cellular Fatty Acid Metabolism and Cancer, Cell
Metabolism, V 18, Aug 2013
9. Dakubo, Mitochondrial Genetics and Cancer, Chapter 2, Springer
2010.
10. Dang, Links between metabolism and cancer, Genes &
Development 26:877–890, 2012
11. DeBerardinis et al, The Biology of Cancer: Metabolic
Reprogramming Fuels Cell Growth and Proliferation, Cell Metabolism 7, January
2008
12. Elliott and Head, Cancer: Tumor Iron Metabolism, Mitochondrial
Dysfunction and Tumor Immunosuppression; “A Tight Partnership—Was Warburg
Correct?”, Journal of Cancer Therapy, 2012, 3, 278-311
13. Elstron, et al, Akt Stimulates Aerobic Glycolysis in Cancer
Cells, Cancer Research 64, 3892–3899, June 1, 2004
14. Giovannucci et al, Diabetes and Cancer, Diabetes Care
33:1674–1685, 2010
15. Herbel et al, Clinical significance of T cell metabolic
reprogramming in cancer, Clin Trans Med (2016) 5:29
16. Hockenberry et al, The Warburg Effect and Beyond: Metabolic
Dependencies for Cancer Cells, in D. E. Johnson (ed.), Cell Death Signaling in
Cancer Biology and Treatment, Cell Death in Biology and Diseases, Springer (New
York) 2013
17. Hsu and Sabatini, Cancer Cell Metabolism: Warburg and Beyond,
Cell, 134, September 5, 2008
18. Serjeant and Dempsey, Ionisation Constants, Pergamon, IUPAC,
(New York) 1979
19. Kim and Dong, Cancer’s Molecular Sweet Tooth and the Warburg
Effect, Cancer Res 2006; 66: (18). September 15, 2006
20. Kroemer and Pouyssegur, Tumor Cell Metabolism: Cancer’s
Achilles’ Heel, Cancer Cell, 13, June 2008
21. Levine and Kutter, The Control of the Metabolic Switch in
Cancers by Oncogenes and Tumor Suppressor Genes, Science, 3 December 2010 Vol
330
22. Liberti and Locasale, The Warburg Effect: How Does it Benefit
Cancer Cells?, Trends in Biochemical Sciences, Cell Press, 2015, http://dx.doi.org/10.1016/j.tibs.2015.12.001
23. Mahan, Elementary Chemical Thermodynamics, Dover (New York)
2006.
24. Marks et al, Cellular Signal Processing, 2nd Edition,
Garland New York) 2017
25. Massie et al, The androgen receptor fuels prostate cancer by
regulating central metabolism and biosynthesis, The EMBO Journal (2011) 30,
2719–2733
26. McGarty, T., Cancer Immunotherapy: A Systems Approach, Draft
Book, 2017 https://www.researchgate.net/publication/314090163_Cancer_Immunotherapy_A_Systems_Approach?_iepl%5BviewId%5D=yKlkSte1HMQl062opMfTdkOD&_iepl%5BprofilePublicationItemVariant%5D=default&_iepl%5Bcontexts%5D%5B0%5D=prfpi&_iepl%5BtargetEntityId%5D=PB%3A314090163&_iepl%5BinteractionType%5D=publicationTitle
27. McGarty, T., Inflammation, The Immune System and Cancer, Working
Paper, 2017 https://www.researchgate.net/publication/320808126_INFLAMMATION_THE_IMMUNE_SYSTEM_AND_CANCER?_iepl%5BviewId%5D=yKlkSte1HMQl062opMfTdkOD&_iepl%5BprofilePublicationItemVariant%5D=default&_iepl%5Bcontexts%5D%5B0%5D=prfpi&_iepl%5BtargetEntityId%5D=PB%3A320808126&_iepl%5BinteractionType%5D=publicationTitle
28. McGarty, T. Metformin, Statins and Prostate Cancer, 2015, http://www.telmarc.com/Documents/White%20Papers/123MetforminStatin.pdf
29. Moore, W., Physical Chemistry, 4th Edition, Prentice
Hall (Englewood Cliffs) 1972
30. Najafov and Alessi, Uncoupling the Warburg effect from cancer,
PNAS | November 9, 2010 | vol. 107 | no. 45 | 19135–19136
31. Patel and Powell, Warburg meets epigenetics, Science, October
2016.
32. Pavlova and Thompson, The Emerging Hallmarks Of Cancer
Metabolism, Cell Metab. 2016 January 12; 23(1): 27–47
33. Peng, et al, Aerobic glycolysis promotes T helper 1 cell
differentiation through an epigenetic mechanism, Science, October 2016.
34. Riccio, P., Cancer Cell Metabolism, NYAS Conference Report,
August 2015, https://www.nyas.org/ebriefings/cancer-cell-metabolism/
35. Seyfried and Shelton, Cancer as a metabolic disease, Nutrition
& Metabolism 2010, 7:7
36. Thompson, C., Wnt meets Warburg: another piece in the puzzle?, The
EMBO Journal, 2014.
37. Tinoco, et al, Physical Chemistry, Prentice Hall (New York)
1995.
38. Tisdale, Mechanisms of Cancer Cachexia, Physiol Rev 89: 381–410,
2009
39. Tran et al, Targeting Cancer Metabolism - Revisiting the Warburg
Effects, Toxicol. Res. Vol. 32, No. 3, pp. 177-193 (2016)
40. Vander Heiden, et al, Understanding the Warburg Effect: The
Metabolic Requirements of Cell Proliferation, Science May 2009Walas, Reaction
Kinetics for Chemical Engineers, McGraw Hill (New York) 1959
41. Warburg O. On the origin of cancer cells. Science 1956 Feb
24;123 (3191):309–14
42. Wittig and Coy, The Role of Glucose Metabolism and
Glucose-Associated Signalling in Cancer, Perspectives in Medicinal Chemistry
2007:1 64–82
43. Xu et al, Warburg Effect or Reverse Warburg Effect? A Review of
Cancer Metabolism, Oncol Res Treat 2015;38:117–122
44. Yun et al, Interactions between epigenetics and metabolism in cancers,
Frontiers in Oncology, Molecular and Cellular Oncology, November 2012, Volume2,
Article 163
45. Zwaans and Lombard, Interplay between sirtuins, MYC and
hypoxia-inducible factor in cancer-associated metabolic reprogramming, Disease
Models & Mechanisms (2014) 7, 1023-1032
[1] Seyfried
and Shelton Nutrition & Metabolism 2010, 7:7 http://www.nutritionandmetabolism.com/content/7/1/7
Page 15 of 22
[2]
Again see Seyfried and Shelton, In addition to avoiding exposure to
established cancer risk factors, the metabolism of ketone bodies protects the
mitochondria from inflammation and damaging ROS. ROS production increases
naturally with age and damages cellular proteins, lipids, and nucleic acids.
Accumulation of ROS decreases the efficiency of mitochondrial energy production.
The origin of mitochondrial ROS comes largely from the spontaneous reaction of
molecular oxygen (O2) with the semiquinone radical of coenzyme Q, .QH, to
generate the superoxide radical O2-. Coenzyme Q is a hydrophobic molecule that
resides in the inner mitochondrial membrane and is essential for electron
transfer. Ketone body metabolism increases the ratio of the oxidized form to
the fully reduced form of coenzyme Q (CoQ/CoQH2). Oxidation of the coenzyme Q
couple reduces the
amount of the semiquinone radical, thus decreasing
superoxide production.
[3]
See Berg, Biochemistry.
[4]
See Rodwell et al, Biochemistry, McGraw Hill (New York) 2915, pp 160-170,
Ferrier, Biochemistry, 6th Edition,
Lippincott (New York) 2014 pp 109-120.
[5]
Details of the TCA can be found in the references.


